雷诺嗪后处理对大鼠心肌再灌注损伤营救激酶及线粒体渗透性转换孔道的影响
2010-12-08陈玉培向许进
陈玉培,向许进,闵 苏
我们在前期试验中发现雷诺嗪后处理(ranolazine postconditioning,RpostC)能改善大鼠离体缺血/再灌注心肌心功能及冠脉流量,降低冠脉流出液中肌酸激酶(CK)与乳酸脱氢酶(LDH)的活性和心肌肌钙蛋白Ⅰ(CTnI)的含量,但其作用机制尚不清楚。研究证实[1],缺血后处理的抗凋亡作用与其抑制线粒体通透性转换孔道(mitochondrial permeability transition pore,MPTP)开放和激活再灌注损伤营救激酶(reperfusion injurysalvage kinase,RISK)途径有关。本研究通过观察雷诺嗪后处理对大鼠离体缺血/再灌注心肌细胞凋亡、MPTP和RISK途径的影响,初步探讨雷诺嗪后处理心肌保护的作用机制。
1 材料与方法
1.1 实验动物 健康清洁级成年SD大鼠,体重180~250 g,由重庆医科大学动物中心提供。
1.2 主要仪器及试剂 离体心脏灌流系统(上海奥尔科特生物科技有限公司),Ultrospec 2100 pro紫外/可见分光光度仪(美国通用电器),IMMULITE 2000化学发光免疫分析仪(天津德普诊断产品有限公司),PowerPac HC电泳仪、GelDoc凝胶成像系统和quantity one分析软件(美国 BIO-RAD)。雷诺嗪(ranolazine,092K4708,美国 Sigma 公司),atractyloside(MPTP 开放剂,029H06461,美国 Sigma),PD98059(ERK1/2抑制剂,24661-02S,美国 Biosource);wortmannin(Wort,PI3K 抑制剂,L15531,美国 Alexis Biochemicals),p-P44/42 MAPK(Thr202/Tyr204)(E10)小鼠单克隆抗体(#9106S,美国Cell Signaling Technology),p-磷 酸 化-AKT(Ser473)(587F11)小鼠单克隆抗体(#4051S,美国Cell Signaling Technology),GAPDH人单克隆抗体(bsh-0510M,北京博奥森生物技术有限公司),辣根酶标记山羊抗小鼠IgG(H+L)(ZB-2305,北京中杉金桥生物技术有限公司)。
1.3 Langendorff心脏灌注模型的建立 SD大鼠腹腔注射 20%乌拉坦 1.0 g·kg-1及肝素钠1 000 U·kg-1,麻醉后无菌下开胸,快速取出心脏,立即置于预先用95%O2+5%CO2混合气体(1.5 L·min-1)饱和的4℃ K-H 缓冲液[成分(mmol·L-1):NaCl 118、KCl 4.7、KH2PO41.2、CaCl22.5、NaHCO325.0、MgSO41.2、葡萄糖 11.1、EDTA-2Na 0.125]中漂洗并轻轻挤出心脏中的血液;用眼科镊将主动脉置于Langendorff灌注管口,用丝线固定后行心脏逆行恒压灌注(灌注压为为100 cm H2O)。整个操作过程应在2 min内完成。K-H缓冲液的灌注压为100 cmH2O、PO266.7 ~73.3 kPa、PCO24.8~5.6 kPa,pH值7.35~7.45。整个灌注系统及心脏周围用恒温水浴循环器维持在(37±0.5)℃,室温控制在(25±1)℃。灌流开始数秒内心脏即可开始跳动。平衡20 min,心脏搏动达到稳定状态后开始进一步实验。
1.4 实验分组与处理 56只成年SD大鼠随机分为8组(n=7):①缺血/再灌注组(I/R组):全心停灌30 min,用K-H液再灌注15 min;② 雷诺嗪后处理组(RPostC 组):全心停灌 30 min,用含 20 μmol·L-1雷诺嗪的K-H液再灌注10 min,再用K-H液灌注5 min;③ 雷诺嗪+atractyloside组(RA组):用含20 μmol·L-1雷诺嗪 +20 μmol·L-1atractyloside的K-H液再灌注10 min;④ 雷诺嗪+PD98059组(RP 组):用含 20 μmol·L-1雷诺嗪 +10 μmol·L-1PD98059的K-H液再灌注10 min;⑤ 雷诺嗪+wortmannin(RW 组):用含 20 μmol·L-1雷诺嗪 +1 μmol·L-1wortmannin的 K-H 液再灌注10 min;⑥atractyloside 组(A 组):用含 20 μmol·L-1atractyloside的K-H液再灌注10 min;⑦ P组:用含10 μmol·L-1PD98059的K-H液再灌注10 min;⑧ W组:用含1 μmol·L-1wortmannin的 K-H 液再灌注 10 min。RA组、RP组、RW组、A组、P组、W组的余处理同RPostC组。
1.5 取材 再灌注末,立即从灌注架上取下心脏,在冰盘中取左心室缺血区组织,将其剪成3块称重。1块用40 g·L-1多聚甲醛固定,检测心肌细胞凋亡,另两块用铝铂纸包好,液氮冻存备用。
1.6 心肌细胞凋亡检测 用TUNEL法。将多聚甲醛固定的心肌组织常规石蜡包埋并作心肌组织切片(5 μm),切片经脱蜡、脱水、DAB显色后,在光镜下随机拍摄5个400倍视野,正常细胞核呈蓝色,细胞核染成棕黄色即为凋亡阳性细胞,计算平均阳性细胞率,用凋亡指数(AI)反映心肌凋亡程度(AI=凋亡阳性细胞数/总细胞数×100)。
1.7 MPTP开放程度检测[2]用分光光度计法。用差速离心法提取心肌组织线粒体,用280 nm波长测线粒体浓度并调整线粒体浓度为0.25 g·L-1。平衡2 min后,540 nm波长测吸光度初值(△A1),加0.25 mmol·L-1CaCl2诱导 MPTP 开放,10 min后吸光度不再变化,记录此时吸光度值(△A2)。用△A=△A1-△A2表示 MPTP开放程度,△A越小,MPTP开放程度越大。
1.8 p-AKT、p-ERK1/2蛋白表达检测 用Western blot法,内参照为GAPDH。用Bradford方法测定心肌细胞蛋白含量,取样品蛋白100μg,经 SDSPAGE电泳、NC转膜、加入一抗(小鼠抗大鼠 p-AKT、p-ERK1/2或GAPDH单克隆抗体,分别按1∶1 000、1 ∶2 000或1 ∶200)室温孵育 2 h,PBS洗 5 min×3次,TBS洗5 min×3次,加入辣根过氧化物酶(HRP)标记的山羊抗小鼠二抗(1∶5 000),室温封闭1 h,TBS洗5 min×3次,化学发光法显色,用GelDoc凝胶成像系统中曝光、成像,Quantity one分析软件分析条带密度值,用p-AKT、p-ERK 1、p-ERK 2密度值分别与GAPDH密度值的比值表示蛋白表达水平。
2 结果
2.1 雷诺嗪后处理对大鼠离体缺血/再灌注心肌细胞凋亡和MPTP开放程度的影响 与IR组比较,RPostC组心肌细胞AI和MPTP开放程度均下降(P<0.05),其余各组差异均无统计学意义(P>0.05);与 RPostC 组比较,RA、RP、RW、A、P 和 W 组AI和MPTP开放程度均升高(P<0.05);与RA组比较,RP、RW、A、P和W组AI和MPTP开放程度差异均无统计学意义(P>0.05),见Tab 1。
Tab 1 Comparison of the AI and the opening of MPTPin each group(¯±s,n=7)
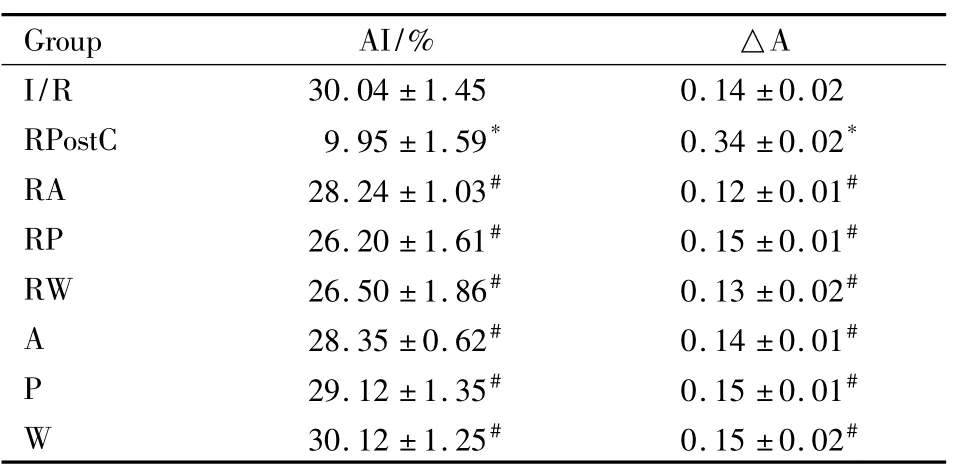
Tab 1 Comparison of the AI and the opening of MPTPin each group(¯±s,n=7)
*P<0.05 vs I/R group;#P<0.05 vs RPostC group
Group AI/% △A I/R 30.04±1.45 0.14±0.02 RPostC 9.95±1.59* 0.34±0.02*RA 28.24±1.03# 0.12±0.01#RP 26.20±1.61# 0.15±0.01#RW 26.50±1.86# 0.13±0.02#A 28.35±0.62# 0.14±0.01#P 29.12±1.35# 0.15±0.01#W 30.12±1.25# 0.15±0.02#
2.2 雷诺嗪后处理对大鼠离体缺血再灌注心肌细胞p-AKT、p-ERK1/2蛋白表达的影响 与IR组比较,RPostC、RA、RP 和 RW 组 p-AKT、p-ERK1/2蛋白表达均升高(P<0.05),A、P和W组差异无统计学意义(P>0.05);与 RPostC 组比较,RP、RW、A、P和W组 p-AKT、p-ERK1/2蛋白表达均下降(P<0.05),RA组差异无统计学意义(P>0.05)。与RA组比较,RP、RW、A、P 和 W 组 p-AKT、p-ERK1/2 蛋白表达均下降(P<0.05),见Tab 2。
Tab 2 The expression of p-AKT and p-ERK1/2 protein in each group(¯±s,n=7)

Tab 2 The expression of p-AKT and p-ERK1/2 protein in each group(¯±s,n=7)
*P<0.05 vs IR group;#P<0.05 RPostC group;▲P<0.05 vs RA group
Group p-AKT p-ERK1 p-ERK2 I/R 0.42±0.04 0.84±0.05 0.87±0.03 RPostC 0.66±0.03* 1.02±0.04* 1.00±0.02*RA 0.67±0.03* 1.05±0.06* 1.00±0.03*RP 0.51±0.03*#▲ 0.94±0.02*#▲ 0.94 ±0.02*#▲RW 0.52±0.04*#▲ 0.93±0.02*#▲ 0.94 ±0.01*#▲A 0.42±0.05#▲ 0.84±0.03#▲ 0.89 ±0.02#▲P 0.41±0.05#▲ 0.85±0.05#▲ 0.89 ±0.03#▲W 0.42±0.04#▲ 0.85±0.04#▲ 0.89±0.02#▲
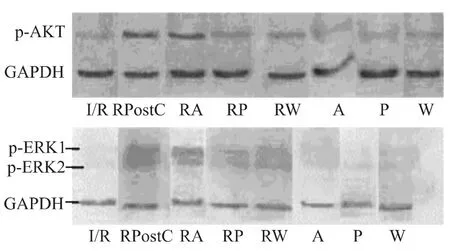
Fig 1 The expression of p-AKT and p-TERK1/2 protein in each group
3 讨论
缺血后处理是具有明确保护效果的一种内源性保护机制[3~5],但其临床应用受限。药物后处理是用药物模拟缺血后处理的方式,模拟或者激发缺血后处理中的心肌保护机制或者一些内源性而呈现与缺血后出相似的保护效果,是临床研究后处理努力的方向。雷诺嗪(ranolazine)是由美国Syntex公司研发[6]的一种不引起血液动力学变化,不引起心率和血压改变的新型抗心绞痛药,其减轻心肌缺血/再灌注损伤作用已有文献报道。我们前期研究证实,雷诺嗪后处理具有明显的心肌保护效应,并发现其发挥最大心肌保护效应的有效浓度为20 μmol·L-1。另有研究表明[7,8],大鼠离体心再灌注 15 min末,p-AKT明显增加,因此,本实验选择雷诺嗪20 μmol·L-1和再灌注15 min分别作为实验浓度和观察时点。本研究发现,雷诺嗪后处理组的心肌细胞凋亡和凋亡指数明显低于I/R组,表明雷诺嗪后处理具有抗心肌细胞凋亡作用,与文献报道一致[9,10]。
MPTP是一个横跨在线粒体内外膜的孔道,在钙浓度升高和氧化应激等因素作用下可以诱导其开放,MPTP开放后,小分子溶质自由通过线粒体内膜,线粒体基质中高浓度大分子蛋白产生胶体渗透压使水进入线粒体,导致线粒体基质肿胀、嵴伸展、外膜破裂,使膜间隙成分如细胞色素C、凋亡诱导因子、Diablo-SMAC等进入胞质,进一步激活caspase-9和 caspase-3,从而诱导细胞凋亡[11,12]。本研究发现,雷诺嗪后处理组较缺血/再灌注组心肌细胞MPTP的开放程度明显降低,心肌细胞凋亡明显减少,而用MPTP开放剂atractyloside干预雷诺嗪后处理后,其MPTP的开放程度明显升高,心肌细胞凋亡也明显增多,提示雷诺嗪后处理抗心肌细胞凋亡作用与其抑制MPTP开放有关。
RISK途径是指在再灌注期间发挥抗细胞死亡心脏保护作用的磷脂酰肌醇3激酶(phosphatidylinositol 3-kinase,P13K)-AKT和细胞外信号调节激酶(extracellular signal-regulated kinase 1/2,ERKl/2)前生存激酶的信号级联[1],是再灌注时重要的心肌保护靶点之一。研究表明[6],缺血后处理可通过激活RISK途径,减轻缺血/再灌注心肌的细胞凋亡。本研究结果表明,雷诺嗪后处理组较缺血/再灌注组p-AKT、p-ERK1/2蛋白表达明显升高,心肌细胞凋亡明显减少,而用 PI3K抑制剂 Wortmmanin和ERK1/2抑制剂 PD9805干预后,p-AKT、p-ERK1/2蛋白表达明显下降,心肌细胞凋亡明显增多,提示雷诺嗪后处理的抗心肌凋亡作用与其进一步激活RISK途径有关。本研究还发现,用MPTP开放剂atractyloside干预雷诺嗪后处理组,其心肌细胞凋亡明显增多,MPTP开放程度增大,但p-AKT、p-ERK1/2蛋白表达水平与雷诺嗪后处理组无差异,提示雷诺嗪后处理的抗心肌凋亡作用可能是先通过激活RISK途径,再进一步抑制MPTP开放实现的,这种作用机制与缺血后处理的RISK-MPTP机制极为类似[6],说明雷诺嗪后处理可以模拟缺血后处理。
综上所述,雷诺嗪后处理对大鼠离体缺血/再灌注损伤心肌具有明显的抗凋亡作用,其机制可能与其激活RISK途径和抑制MPTP开放有关。
[1] Hausenloy D J,Yellon D M.New directions for protecting the heart against ischaemia-reperfusion injury:targeting the reperfusion injury salvage kinase(RISK)-pathway[J].Cardiovasc Res,2004,1561(3):448-60.
[2] Rajesh K G,Sasaguri S,Suzuki R,et al.Antioxidant MCI-186 inhibits mitochondrial permeability transition pore and upregulates Bcl-2 expression[J].Am J Physiol Heart Circ Physiol,2003,285(5):H2171-8.
[3] 吾艳娜,娄建石.心肌缺血预适应和后适应在再灌注期共同靶点的研究进展[J].中国药理学通报,2007,23(8):993 -6.
[3] Wu Y N,Lou J S.Advances in study of common targets of myocardial ischemic preconditioning and postconditioning during reperfusion[J].Chin Pharmacol Bull,2007,23(8):993 -6.
[4] 李敬远,王俊科,曾因明.外周苯二氮艹卓受体参与调控大鼠心肌线粒体通透性转换[J].中国药理学通报,2007,23(3):341-5.
[4] Li J Y,Wang J K,Zeng Y M.Involvement of peripheral benzodiazepine receptor in the regulation of rat cardiac mitochondria permeability transition[J].Chin Pharmacol Bull,2007,23(3):341 -5.
[5] 任静华,陈玉培.缺血后处理对大鼠离体心脏缺血/再灌注损伤的作用[J].中华麻醉学杂志,2007,27(1):84 -7.
[5] Ren J H,Chen Y P.Effects of ischemic postconditioning on myocardial ischemic-reperfusion injury to isolated rat heart[J].Chin J Anesthesiol,2007,27(1):84 -7.
[6] Hausenloy D J,Tsang A,Yellon D M.The reperfusion injury salvage kinase pathway:a common target for both ischemic preconditioning and postconditioning[J].Trends Cardiovasc Med,2005,15(2):69-75.
[7] Feng J,Lucchinetti E,Ahuja P,et al.Isoflurane postconditioning prevents opening of the mitochondrial permeability transition pore through inhibition of glycogen synthase kinase 3beta[J].Anesthesiology,2005,103(5):987 -95.
[8] Schulman D,Latchman D S,Yellon D M.Urocortin protects the heart from reperfusion injury via upregulation of p42/p44 MAPK signaling pathway[J].Am J Physiol Heart Circ Physiol,2002,283(4):H1481-8.
[9] Sun H Y,Wang N P,Halkos M,et al.Postconditioning attenuates cardiomyocyte apoptosis via inhibition of JNK and p38 mitogen-activated protein kinase signaling pathways[J].Apoptosis,2006,11(9):1583-93.
[10]Cserepes B,Jancsó G,Gasz B,et al.Cardioprotective action of urocortin in early pre-and postconditioning[J].Ann N Y Acad Sci,2007,1095:228 -39.
[11]Krolikowski J G,Bienengraeber M,Weihrauch D,et al.Inhibition of mitochondrial permeability transition enhances isoflurane-induced cardioprotection during early reperfusion:the role of mitochondrial KATPchannels[J].Anesth Analg,2005,101(6):1590 -6.
[12]Bernardi P,Rasola A.Calcium and cell death:the mitochondrial connection[J].Subcell Biochem,2007,45:481 -506.
