肾小球滤过屏障的新理念
2010-11-26综述郑春霞审校
汤 曦 综述 郑春霞 审校
传统观点认为肾小球滤过屏障(glomerular filtration barrier,GFB)由内皮细胞、肾小球基膜(glomerular basement membrane,GBM)以及足细胞三层结构组成。最新发现内皮细胞表层(endothelial surface layer,ESL)和足细胞下间隙(subpodocyte space,SPS)均具有溶质分子筛选特征,对肾小球滤过功能具有重要影响。因此,本文就肾小球滤过屏障五层结构(图1)及相互调节的研究进展进行综述。
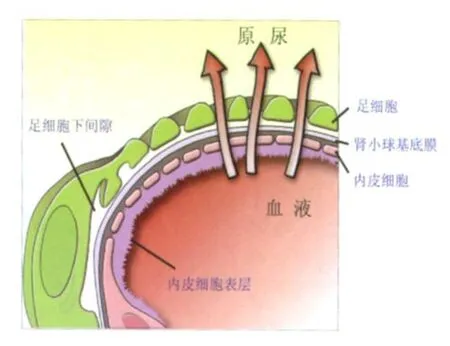
图1 肾小球滤过屏障五层结构[1]
肾小球的选择通透性
GFB的功能主要体现在对水和小分子溶质的自由通透性和对中大分子溶质的选择通透性。大量证据支持 GFB对白蛋白等溶质分子具有高度选择通透性。GFB对溶质分子的选择性常用筛选系数 θ来评价。θ=某溶质包曼囊内浓度/相应的血浆浓度。白蛋白等带有负电荷的大分子主要以弥散方式通过 GFB。按已知的多层滤过屏障模型分析,θ为各层滤过屏障筛选系数之积,且各层筛选系数间相互影响。θ为复合参数,受两方面因素影响:一方面为溶质的浓度、电荷、大小、通过 GFB的方式以及GFB表面作用力如毛细血管压等;另一方面为 GFB本身对毛细血管内物质的滤过阻力,由水传导率(水自由通过 GFB的能力)、溶剂曳引反射系数 (水流曳引溶质跨过 GFB的程度)以及溶质弥散通透力(溶质弥散的阻力大小)构成[1]。后者中各参数值的大小取决于 GFB的精细结构。因此,对 GFB各层结构和功能的深入认识对了解肾小球整体滤过功能具有重要意义。
肾小球内皮细胞表层
肾小球内皮细胞表层是指覆盖于肾小球内皮细胞、窗孔表面的凝胶状糖萼以及附着的部分血浆蛋白[2]。内皮细胞表层糖萼为富含碳水化合物的网状结构,由带阴电荷的蛋白核心 (perlecan、syndecan、versican)与侧链糖胺聚糖(硫酸肝素,硫酸软骨素)共价结合形成。这些组分由内皮细胞自身合成分泌,并结合于细胞膜表面或填充于内皮窗孔间形成网塞[3]。ESL厚度约 200~400 nm[4],常规电镜处理标本后需再加阳离子染色才能显示。由于组织块经固定、脱水等技术处理可能导致 ESL塌陷,所以测得的 ESL厚度常小于实际厚度。
ESL组分的排列和密度决定了筛选分子的范围及静水压阻力[5],构成了肾小球滤过的第一道屏障。体外实验发现 ESL部分降解酶(如肝素酶、硫酸软骨素酶、玻璃酸酶)可导致离体灌注肾白蛋白通透性增加[6]。Singh等[7]进一步采用神经氨酸酶作用于永生化温度敏感人肾小球内皮细胞,在排除ESL降解酶对 GBM和足细胞表面分子影响的情况下,发现内皮细胞白蛋白通透率及电荷阻力均发生变化。硫酸肝素蛋白聚糖(heparan sulfate proteoglycan,HSPG)经肝素酶 III或人肝素酶选择性裂解后,白蛋白跨内皮细胞的通透性增加,而电荷阻力无变化。这提示 ESL特别是 HSPG在肾小球滤过中起着重要作用。在阿霉素肾病小鼠模型中发现肾小球ESL较正常对照组减少 80%,白蛋白通透性增加[8]。
此外,ESL附着的血清粘蛋白、白蛋白等血浆成分,与血管内皮细胞表面的糖萼发生作用,参与调节毛细血管的通透性[9]。血清粘蛋白为高度唾液酸化、多价阴离子的糖蛋白,由肝脏或毛细血管内皮细胞合成分泌,并与 ESL糖萼结合,加强阴离子电荷屏障,降低毛细血管通透性[10,11]。遗传性白蛋白缺陷大鼠尿液中 60~90 kD大小的蛋白分子增加,注射小牛血清白蛋白 12h后尿蛋白消失[12]。这提示循环中的白蛋白分子参与维持肾小球选择通透性。
肾小球内皮细胞
肾小球毛细血管内皮细胞属窗孔型内皮细胞。肾小球内皮窗孔总面积占内皮细胞表面积的 20%~50%;孔径约 60~100 nm,为白蛋白分子直径(3.6 nm)的 15倍以上[14],水和小分子溶质易通过,但不能限制中大分子物质通过。
肾小球内皮窗孔是否存在隔膜一直为研究的焦点。Rostgaard等[15]发现窗孔内存在丝状网塞,即富含硫酸肝素、透明质酸的 ESL糖萼[14,16],能阻止大分子物质通过。Hjalmarsson等采用还原铁氰化物锇酸染色法,电镜下显示了窗孔内有 1或 2层隔膜[4]。据此,人们推测窗孔隔膜可能支撑上层的ESL并调节肾小球窗孔直径。但并非所有的肾小球内皮窗孔均有隔膜。肾小球内皮细胞窗孔的发育形成及由隔膜型转化为无隔膜型的过程受血管内皮生长因子(vascular endothelial growth factor,VEGF)调节[14]。Ichimura等发现成熟小鼠的肾小球中仅 2%毛细血管内皮细胞窗孔表达细胞膜囊泡相关蛋白 1(plasmalemmal vesicle-associated protein-1,PV-1)[17]。有趣的是,在肾小球发育及 Thy-1.1肾炎恢复的不同阶段,肾小球内皮窗孔 PV-1的表达不一。未成熟的肾小球和治疗后的 Thy-1.1肾炎中,内皮窗孔隔膜数目和 PV-1表达增加[18]。这一方面提示随着肾小球分化发育成熟,隔膜结构逐渐消失,以保证高效滤过功能;另一方面,肾小球疾病恢复过程中,窗孔隔膜及 PV-1的重现可能参与了肾小球毛细血管内皮的重塑修复过程。
肾小球基膜(GBM)
GBM为凝胶状的细胞外基质,主要由 IV型胶原、层粘连蛋白(laminin)、巢蛋白以及蛋白聚糖构成,为最重要的静水压缓冲结构。随着 GBM的发育成熟,IV胶原的组分由 α1、α2亚链逐渐向更加坚韧的 α3、α4、α5亚链转化。发育成熟的 GBM中laminin构成以 α5β2γ1链为主,链接 GBM各成分,并与足细胞基底侧表面受体 α3β1整合素、α6β1整合素及 α-dystroglycan相互作用。GBM中蛋白聚糖以 HSPG为主,主要分布于外疏松层。GBM中HSPG的基质蛋白核心包括基膜蛋白聚糖、集聚蛋白(agrin)、胶原 XVIII。随着肾单位的发育,GBM中基膜蛋白聚糖、胶原 XVIII减少,而 agrin表达增加,构成 GBM中主要的 HSPG。
GBM既往被认为是肾小球滤过的主要机械和电荷屏障。随着足细胞裂孔膜复合体的发现,人们认为 GBM是肾小球的粗过滤装置。部分学者认为与 agrin相连的 HSPG是 GBM电荷屏障的主要组分,HSPG的减少是蛋白尿产生的原因。动物实验发现非肥胖性糖尿病小鼠、嘌呤霉素肾病、被动型Hyemann肾炎模型中均有硫酸肝素表达下调。Raats等[19]利用 agrin抗体和硫酸肝素抗体观察不同肾脏疾病患者的 GBM,发现 agrin没有变化,侧链硫酸肝素表达量与蛋白尿呈反比。并探讨了不同肾脏疾病动物模型中硫酸肝素减少的机制:狼疮性肾小球肾炎中抗核抗体核小体遮蔽了硫酸肝素;阿霉素肾病中硫酸肝素被活性氧自由基解聚;链脲佐菌素(streptozotocin,STZ)大鼠中高糖抑制硫酸肝素的合成。Van den Hoven等[20]也发现尿蛋白 >2g/24h的显性糖尿病肾病(DN)患者中硫酸肝素表达减少,伴有肝素酶的升高。相反,有的学者认为硫酸肝素减少可能是继发于蛋白尿的变化。因为离体GBM对阴电荷硫酸蔗聚糖和中性蔗聚糖的筛选系数无差异;早期 DN和微小病变性肾病患者的 GBM硫酸肝素表达无明显变化[21,22];体内注射类肝素酶的小鼠未产生蛋白尿[23];足细胞特异 Ext1基因敲除的小鼠缺乏硫酸肝素侧链多聚化相关的糖基转移酶,出生 8月后才产生蛋白尿[24]。然而,肝素酶抗体或抑制剂 PI88可以减少被动型 Hyemann肾炎大鼠的蛋白尿[25]。综上可见,HSPG在肾脏蛋白尿的发生中并非仅仅依赖于其阴电荷屏障作用。因为硫酸肝素还介导足细胞与基膜黏附、结合并转导生长因子信号等[19]。此外,IV型胶原 α3和(或)α5亚链缺失所致的 Alport综合征,及近期研究发现lamininβ2、α5缺陷导致的蛋白尿[26,27],均提示 GBM为肾小球机械屏障的重要组分。
足细胞与裂孔膜复合体
蛋白尿的产生常伴有足突融合等足细胞损伤现象。1999年nephrin的发现,让人们认识到足细胞和裂孔膜复合体是 GFB的最关键部分。随后发现许多足细胞分子基因突变导致大量蛋白尿,如nephrin(NPSH1)、podocin(NPHS2)、α-actinin IV(ACTN4)、瞬时受体电位阳离子通道蛋白 6(transient receptor potential cation channel 6,TRPC6)、willims瘤抑癌基因 (WT1)、lamininβ2(LAMB2)、Phospholipase C eplison 1(PLCE1)。根据分布的部位,足细胞分子可以分为裂孔膜、顶膜、基底膜及骨架蛋白四大类。其中细胞骨架蛋白为中心环节,其余三类分子直接或间接与骨架蛋白相连,动态维持足细胞的极性结构和功能。
足细胞与神经元类似,为高度分化的极性细胞,通过极性复合物与小 G蛋白如 Rac、Rho、Cdc42协调作用,调节细胞骨架,维持顶部、底部和侧部分子的分布及功能差异。目前已知在上皮细胞极性建立和维持中最主要的三个极性复合物是:Par-aPKC复合物、Scribble(Lgl-Dlg-Scrib)复合物和 Crb(Crb-Pals-PATJ)复合物。三者共同配合发挥功能。Par-aPKC复合物即 aPKC-Par3-Par6,分布于上皮细胞紧密连接处、神经元轴突生长锥和足细胞裂孔膜。Par-aPKC复合物在足细胞分化发育过程中由顶部转至基底面和侧面,早于裂孔膜复合体的形成,在维持足突的形态功能以及诱导足细胞损伤后修复中起着重要作用。Par3为载体蛋白,承载 Par6和蛋白激酶 C(aPKC),与 Par6同样具有 PDZ结构域,即最初发现于后突触密度蛋白(PSD95/SDP90)、果蝇肿瘤抑制因子(DLG-A)和紧密连接蛋白(ZO-1)中 80~90 kD大小的同源结构。Par3的两个 PDZ结构域分别直接与 nephrin以及 neph1结合,参与足细胞裂孔膜复合体的信号转导。Rac、Cdc42激活后导致 Par6构象改变,促进 aPKC磷酸化激活。活化后的 aPKC直接结合并磷酸化 KIBRA(kidney brain)。KIBRA为新发现的一种具有 WW结构域的胞浆蛋白,因主要分布于大脑和肾脏而得名,在肾脏正常表达于足细胞、部分小管及集合管上皮细胞内。足细胞内的KIBRA与 PATj作用后结合dendrin、synaptopodin,调节肌动纤维,使足细胞定向移行[28]。抑制 aPKC可致足突融合和细胞骨架紊乱;足细胞特异性敲除aPKCλ/ι的小鼠足细胞 Podocalyxin、裂孔膜分子分布异常伴毛细血管袢节段硬化,表现为肾病综合征[29,30]。
足细胞骨架结构的动态调节在维持 GFB功能中的地位越来越被重视。体外永生化温度敏感小鼠和人足细胞系的建立使足细胞信号通路的研究成为可能。 nephrin、podocin、α3β1整合素等分子除作为足细胞结构分子外,还具有跨膜信号转导作用。近期在获得性肾脏疾病中,参与足细胞损伤的受体分子及信号通路的相关研究取得了突破性进展。尿激酶受体(urokinase receptor,uPAR)、M型磷脂酶 A2受体(phospholipase A2 receptor,PLA2R)Notch、钙调神经蛋白(calcineurin)等分子的发现为足细胞病变的发病机制以及针对性治疗开启了新纪元。
uPAR为分布广泛的细胞表面糖基磷脂酰肌醇锚定蛋白激酶,无胞内结构域,通过结合尿激酶或与整合素、G蛋白偶联受体等分子作用转导信号,调节细胞流动性,参与炎症、损伤修复以及肿瘤转移等。近来 Wei等[31]发现 uPAR-αvβ3-Vitronectin信号通路参与了蛋白尿的产生。uPAR在正常肾组织固有细胞中组成性表达,而高表达于 DN、局灶节段性肾小球硬化 (FSGS)患者的足细胞。脂多糖(lipopolysaccharide,LPS)诱导小鼠足细胞 uPAR上调、Vitronectin表达增加,激活 β3整合素,导致蛋白尿;而 uPAR基因敲除的 Plaur-/-小鼠对 LPS抵抗,无蛋白尿产生。可见,足细胞 uPAR是参与蛋白尿产生的关键分子之一。
PLA2R为 1型跨膜受体,属哺乳动物甘露糖家族,与分泌型的磷脂酶 A2具有高亲和力。人PLA2R最早从肾组织中克隆发现。Beck等[32]采用移植供肾的肾小球与膜性肾病(MN)患者血清反应后进行 western blot和质谱分析,发现 70%特发性膜性肾病患者肾小球足细胞表达 M型 PLA2R;且循环中存在 IgG4亚型的抗 M型 PLA2R抗体,随病情缓解滴度降低。但足细胞 M型 PLA2R介导的胞内信号通路以及对肾小球滤过功能的影响尚待进一步探讨。
Notch是一种跨膜蛋白,由胞外受体和胞内段两部分构成,分为 1-4亚型,表达分布具有组织和细胞特异性。相邻细胞间通过 Notch受体放大并固化彼此间的分子差异,在发育过程中对细胞的分化方向、增殖和凋亡至关重要,决定着细胞的命运[39]。Notch信号通路最初发现于果蝇的神经系统,在神经系统发育的早期通过抑制原神经基因决定外胚祖细胞向具有神经系统或上皮层潜能的干细胞分化,随后进一步调控神经干细胞向中枢神经系统的神经母细胞或周围神经系统的感觉器官前体细胞等特定的神经细胞亚群分化[40]。Notch胞外受体与 Jagged及 Delta家族结合后,经解离素金属蛋白酶(a disintegerin and metalloproteinase,ADAMs)、γ分泌酶复合体裂解产生 Notch胞内结构域(ICN1)。ICN1入核后与转录激活因子 RBPj、MAML、P300结合后诱导 hes、hey家族基因转录。Notch信号通路与肾脏发育和疾病密切相关。最早肾单位囊泡期表达Notch 1和 Notch 2 mRNA,S期内皮细胞和远端小管上皮细胞分别表达 Notch3和 Notch4。Niranjan等[41]发现活性 Notch即 ICN1在健康啮齿类动物中无表达;而在 8周龄的 STZ大鼠、db/db小鼠足细胞中高表达,导致 nephrin、podocin和 WT-1减少,足突融合、足细胞脱落和球性硬化。足细胞特异的 RBPj敲除可以缓解 db/db小鼠肾脏损伤,γ分泌酶抑制剂能减轻嘌呤霉素大鼠的蛋白尿。进一步研究发现DN、FSGS患者中足细胞 ICN1表达上调,激活 p53,介导足细胞凋亡。
calcineurin为近期发现的与足细胞骨架调节密切相关的分子之一。calcineurin是一种钙离子依赖的丝/苏氨酸磷酸激酶,由活性亚基 A(CnA)、催化亚基 B(CnB)构成。A亚基具有 α(CnAα)、β(CnAβ)、γ(CnAγ)三种异构体,分布具有组织特异性。caclineurin最初在大脑发现,通过活化 T细胞核因子 (nuclear factor of activated T-cells,NFAT)信号通路或者与细胞骨架蛋白结合,参与神经系统发育、神经元的轴突延伸和凋亡等[33]。小鼠肾小管表达 CnAα、CnAβ[34],参与肾脏发育、调节肾小管上皮细胞 Na+/K+ATP酶活性和水通道蛋白的表达分布[35,36]。Faul等[37]发现 CnA与 synaptopodin结合,使 synaptopodin去磷酸化、与 14-3-3蛋白解离,从而易被 capthesin L降解,导致肌动蛋白排列紊乱。转基因或 LPS诱导小鼠足细胞 calcineurin表达上调,synaptopodin、RhoA、dynamin、ZO-1表达下降,产生大量蛋白尿。刘志红等[38]研究表明,部分特发性膜性肾病患者的足细胞 CnAα表达增加伴synaptopodin表达减弱。其中 2例患者经 FK506治疗达临床完全缓解后重复肾活检,发现足细胞CnAα表达减少伴随 synaptopodin表达增加。这为MN足细胞损伤的机制以及分子靶向治疗提出了新的阐释和方向。
足细胞下间隙
传统认为肾小球只有两个囊腔即毛细血管腔和包曼囊腔。毛细血管腔内液体通过内皮细胞、GBM,经足细胞裂孔膜滤过后进入包曼囊腔。然而,足细胞发出初级和次级突起,以足突附着于基膜[42],而非以胞体直接锚定。因此,人们推断 GBM与足细胞胞体间可能存在间隙。随着电镜技术的应用,20世纪 50年代人们发现肾小球毛细血管与足细胞胞体间存在腔隙[43]。2005年Neal等采用电镜三维重建技术,进一步证实了足细胞胞体、足突、和基膜三者间存在第三腔隙,即 SPS(图2),占 GBM外表面积的60%[44]。液体经GBM滤过后大部分汇入 SPS,再经狭窄的 SPS出孔,汇入足细胞间腔隙,最后到达包曼囊腔;仅小部分经裂孔膜滤过直接进入包曼囊腔。
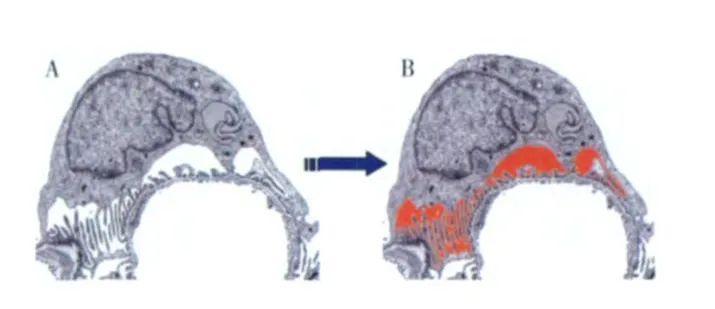
图2 足细胞下间隙[71]
SPS为 GFB阻力的重要组成部分。SPS覆盖区域的毛细血管外周阻力为 SPS裸露区的 900~14 500倍[45]。SPS内的液压为内皮细胞和 GBM承受的水流压力之和的 2.5倍,受SPS出孔调节,在毛细血管外压调节中起着重要作用[45]。足细胞可通过 SPS感受肾小球灌注压和滤过速率,调节整个滤过屏障的阻力。当肾小球灌注压升高时,足细胞通过牵张感受器激活 Ca2+通道,足细胞骨架收缩,SPS出孔缩小,SPS深度缩小且压力增加,限制液流通过GBM[44,45]。
SPS不仅调节滤过屏障液流阻力,还限制中大溶质分子的移动。Salmon等[46]在离体灌注肾小球及大鼠肾脏中,分别选取罗丹明标记的右旋糖酐(10kD)和荧光黄(lucifer yellow)作为示踪剂,采用多光子荧光成像法动态观察比较了不同大小的溶质分子通过肾小球毛细血管袢SPS覆盖区与裸露区的过程。该研究结果发现罗丹明标记的右旋糖酐在肾小球毛细血管腔内灌注浓度稳定的情况下,随着时间推移,肾小球毛细血管腔外围 SPS裸露区荧光强度恒定,而 SPS覆盖区荧光强度逐渐增加。但荧光黄在肾小球毛细血管袢SPS覆盖区和裸露区的荧光强度均恒定,不随时间延长而增加。这提示罗丹明标记的右旋糖酐滞留并蓄积于 SPS,SPS对大分子物质具有筛选作用。
GFB结构间的交互作用
GFB各层之间紧密联系,相互依赖,维持着肾小球的选择通透性。VEGF、骨形成蛋白 (bone morphogenetic protein,BMP)、内皮素 1(endothelin 1,ET-1)、谷氨酰胺等生长因子或神经介质,通过旁分泌或自分泌方式参与 GFB的动态调节。
足细胞与内皮细胞 肾脏局部 VEGF主要由足细胞合成分泌,部分可与硫酸肝素结合贮存于GBM。VEGF以旁分泌或自分泌方式作用于内皮细胞、足细胞本身,在 GFB功能维持中起着重要作用。VEGF家族包括 VEGF-A、B、C、D、E五种类型及胎盘生长因子、血小板衍生生长因子。现已知足细胞合成 VEGF-A、C。肾脏 VEGF-A主要有 VEGF165,120,188三种亚型,以 VEGF165异构体为主,通过VEGF受体 2发挥作用。VEGF-A旁分泌作用于内皮细胞 flt-1、flk-1受体,调节内皮窗孔形成,维持内皮细胞对大分子物质的通透[47,48],而 VEGF-C限制大分子物质通过[49]。自分泌的 VEGF-A、C通过nephrin、PI3K/AKT、p38MAPK等信号通路调节足细胞的存活和功能。VEGF-A的中和抗体通过抑制PI3K/AKT、上调 p38MAPK促足细胞凋亡,而该作用可被外源性 VEGF-C部分缓解[50,51]。缺氧、高糖及血管紧张素 II等刺激 VEGF合成[52]。DN患者早期VEGF升高伴高滤过[53],随着病情进展、球性硬化,VEGF表达减少[54]。先兆子痫患者 sflt-1增加伴NO合成减少,VEGF活性降低,肾脏表现为内皮细胞溶解、血栓性微血管病变[55]。另外,肾小球发育亦受 VEGF的精细调节。足细胞特异敲除 VEGF的小鼠内皮细胞移行、分化、窗孔形成障碍,围产期肾功能衰竭导致死亡[56]。VEGF单个等位基因缺失的小鼠内皮细胞溶解,出生后 2.5周内出现蛋白尿,进行性肾功能恶化,于 9~12周时死亡[56]。而VEGF过度表达的小鼠肾小球毛细血管袢溶解,产生蛋白尿,出生后 1周内肾衰竭死亡[56]。
足细胞产生的 BMP,也通过旁分泌或自分泌方式作用于肾小球内皮细胞或足细胞,维持 GFB的结构和功能。BMP属转化生长因子 β(TGF-β)超家族,与细胞膜上 BMP I型或 II型受体结合后,经Smad或非 Smad(如 p38 MAPK)胞内信号转导,与肾脏、心脑血管疾病密切相关[57]。内皮细胞表面存在 BMP2,4,6,7对应的受体。不同的受体亚型作用各异。BMP2促进内皮细胞移行,BMP4促凋亡,二者均可抑制血管内皮细胞增殖[57]。BMP7为保护性分子,在 DN足细胞中表达减少伴足细胞脱落;转基因或者外源性BMP7通过稳定 podocin、synaptopodin缓解高糖对足细胞的损伤[58]。另外,肾小管上皮细胞产生的 BMP7通过拮抗 TGF-β1抑制肾小管上皮-间充质转分化,延缓糖尿病小鼠肾脏纤维化[59]。
病理情况下,内皮细胞释放的 ET-1激活足细胞内皮素 A受体(endothelin receptor A,ETAR),通过基质金属蛋白酶 9(MMP-9)、p38MAPK、Rho/PI3K等信号通路介导足细胞损伤,导致蛋白尿和肾小球硬化。Collino等[60,61]体外实验发现先兆子痫患者的血清或持续高糖刺激内皮细胞产生ET-1;ET-1作用于足细胞,使 nephrin、synaptopodin表达减少,细胞骨架紊乱。以上病变可被 ETAR阻断剂抑制。临床试验也证实 ETAR阻断剂可以缓解蛋白尿,延缓慢性肾脏疾病进展[62]。此外,内皮细胞损伤后释放的血管紧张素 II、人单核细胞趋化蛋白 1(monocyte chemotactic protein-1,MCP-1),也可以导致足细胞骨架和 nephrin改变[62]。综上可见,无论生理还是病理情况下,肾小球足细胞与内皮细胞间存在紧密联系。
足细胞与基膜 足细胞与 GBM的交互作用对维持 GFB功能至关重要。发育成熟的肾小球中,主要由足细胞合成分泌 GBM成分、基质降解酶,调节GBM的更新。足细胞基底侧膜受体分子α-dystroglycan、整合素与 laminin、agrin结合,介导足细胞黏附于 GBM,并且通过整合素连接激酶(integrin-linked kinase,ILK)等参与信号转导,调节足细胞骨架、足突分化和上皮-间充质转分化。ILK不仅连接整合素与细胞骨架、裂孔膜分子nephrin[63],而且还通过磷酸化下游的 PKB/AKT、GSK-3β等分子,促进足细胞凋亡、β-catenin蓄积并向核内转移,从而诱导 E钙粘素、MMP-9表达,导致足细胞表型转分化、与基膜黏附减弱。因而,ILK被认为是蛋白尿发生中的下游效应分子。DN、先天性芬兰型及移植后复发的 FSGS患者足细胞均存在ILK过度表达[64-66]。ILK抑制剂可阻止足细胞发生上皮-间充质转分化,从而缓解蛋白尿[67]。
GBM与足细胞相辅相成。一方面,基膜分子缺失导致足细胞黏附、分化功能障碍。lamininβ2-/-的小鼠 GBM超微结构似乎完整,但 laminin阴电荷分布异常;出生后早期即产生白蛋白尿,而后出现足突融合和裂孔膜缺失,表现为先天性肾病综合征伴视网膜、神经肌接头病变[68]。常染色体隐性LAMB2突变所致 lamininβ2缺失可致先天性Pierson综合征[26]。另一方面,足细胞病变导致基膜成分改变。足细胞特异的 lamininα5缺失导致基膜虫蚀状增厚,足突融合[27]。LAMA5突变的小鼠发生蛋白尿、血尿和多囊肾,出生 3~4周后死亡[27]。此外,足细胞还通过牵张感受器感受张力,足突收缩调节SPS压力和 GBM的扩张度。
足细胞间突触传递对话 足细胞间的谷氨酰胺信号通路在 GFB中亦起着重要作用。Rastaldi等[69]发现成熟的足细胞具有与神经元类似的谷氨酰胺突触囊泡结构,并且表达与谷氨酰胺胞吐密切相关的小 G蛋白 Rab3A;同时检出肾小球足细胞表达代谢型和离子型谷氨酰胺受体如 N-甲基-D-天冬氨酸受体(N-methyl-D-aspartate receptor,NMDAR)。NMDAR拮抗剂可导致细胞骨架重排、nephrin重分布,而 NMDAR激动剂可以逆转该现象[70]。足细胞特异 Rab3A敲除的小鼠足突融合;特异性阻断NMDAR导致小鼠尿白蛋白增加[70]。临床也观察到采用氯胺酮全身麻醉的患者出现白蛋白尿的现象。可见,足细胞的谷氨酰胺信号通路在维持自身结构和功能中起着重要作用。
展 望
蛋白尿是肾脏疾病的标志,其发生机制一直为研究的焦点。随着分子生物学、免疫学以及电镜三维技术的发展,ESL、SPS的发现,更新了人们对 GFB精细结构的认识。滤过屏障各层结构间的相互调节,在维持固有滤过屏障的功能中起着关键作用。足细胞特异基因的发现,体外永生化温度敏感小鼠和人足细胞系的应用,极大地帮助人们探索足细胞分子的结构和功能。足细胞中尿激酶受体、Notch等分子信号通路的发现,为足细胞靶向性治疗提供了新方向。
1 Haraldsson B,Nyström J,Deen WM.Properties of the glomerular barrier and mechanisms of proteinuria.Physiol Rev,2008,88(2):451-487.
2 Avasthi PS,Koshy V.The anionic matrix at the rat glomerular endothelial surface.Anat Rec,1988,220(3):258-266.
3 Sörensson J,Björnson A,Ohlson M,et al.Synthesis of sulfated proteoglycans by bovine glomerular endothelial cells in culture.Am J Physiol Renal Physiol,2003,284(2):F373-380.
4 Hjalmarsson C,Johansson BR,Haraldsson B.Electron microscopic evaluation of the endothelial surface layer of glomerular capillaries.Microvasc Res,2004,67(1):9-17.
5 Michel CC,Curry FE.Microvascular permeability.Physiol Rev,1999,79(3):703-761.
6 Jeansson M,Haraldsson B.Glomerular size and charge selectivity in the mouse after exposure to glucosaminoglycan-degrading enzymes.JAm Soc Nephrol,2003,14(7):1756-1765.
7 Singh A,Satchell SC,Neal CR,et al.Glomerular endothelial glycocalyx constitutesa barrier to protein permeability.J Am Soc Nephrol,2007,18(11):2885-2893.
8 Jeansson M,Björck K,Tenstad O,et al.Adriamycin alters glomerular endothelium to induce proteinuria.J Am Soc Nephrol,2009,20(1):114-122.
9 Schnitzer JE,Carley WW,Palade GE.Specific albumin binding to microvascular endothelium in culture.Am J Physiol,1988,254(3 Pt 2):H425-437.
10 Schnitzer JE,Pinney E.Quantitation of specific binding of orosomucoid to cultured microvascular endothelium:role in capillary permeability.Am JPhysiol,1992,263(1 Pt 2):H48-55.
11 Sörensson J,Matejka GL,Ohlson M,et al.Human endothelial cells produce orosomucoid,an important component of the capillary barrier.Am J Physiol,1999,276(2 Pt 2):H 530-534.
12 Fujihara CK,Arcos-Fajardo M,Brandão De Almeida Prado E,et al.Enhanced glomerular permeability to macromolecules in the Nagase analbuminemic rat.Am J Physiol Renal Physiol,2002,282(1):F45-50.
13 Bulger RE,Eknoyan G,Purcell DJ 2nd,et al.Endothelial characteristics of glomerular capillaries in normal,mercuric chloride-induced,and gentamicin-induced acute renal failure in the rat.J Clin Invest,1983,72(1):128-141.
14 Satchell SC,Braet F.Glomerular endothelial cell fenestrations:an integral component of the glomerular filtration barrier.Am J Physiol Renal Physiol,2009,296(5):F947-956.
15 Rostgaard J,Qvortrup K.Electron microscopic demonstrations of filamentous molecular sieve plugs in capillary fenestrae.Microvasc Res,1997,53(1):1-13.
16 Rostgaard J,Qvortrup K.Sieve plugs in fenestrae of glomerular capillaries—site of the filtration barrier?Cells Tissues Organs,2002,170(2-3):132-138.
17 Ichimura K,Stan RV,Kurihara H,et al.Glomerular endothelial cells form diaphragms during development and pathologic conditions.JAm Soc Nephrol,2008,19(8):1463-1471.
18 Kriz W.Fenestrated glomerular capillaries are unique.J Am Soc Nephrol,2008,19(8):1439-1440.
19 Raats CJ,Van Den Born J,Berden JH.Glomerular heparan sulfate alterations:mechanisms and relevance for proteinuria.Kidney Int,2000,57(2):385-400.
20 van den Hoven MJ,Rops AL,Bakker MA,et al.Increased expression of heparanase in overt diabetic nephropathy.Kidney Int,2006,70(12):2100-2108.
21 van den Born J,Pisa B,Bakker MA,et al.No change in glomerular heparan sulfate structure in early human and experimental diabetic nephropathy.JBiol Chem,2006,281(40):29606-29613.
22 Wijnhoven TJ,Geelen JM,Bakker M,et al.Adult and paediatric patients with minimal change nephrotic syndrome show no major alterations in glomerular expression of sulphated heparan sulphate domains.Nephrol Dial Transplant,2007,22(10):2886-2893.
23 Wijnhoven TJ,Lensen JF,Wismans RG,et al.In vivo degradation of heparan sulfates in the glomerular basement membrane does not result in proteinuria.J Am Soc Nephrol,2007,18(3):823-832.
24 Chen S,Wassenhove-McCarthy DJ,Yamaguchi Y,et al.Loss of heparan sulfate glycosaminoglycan assembly in podocytes does not lead to proteinuria.Kidney Int,2008,74(3):289-299.
25 Levidiotis V,Freeman C,Punler M,et al.A synthetic heparanase inhibitor reduces proteinuria in passive Heymann nephritis.J Am Soc Nephrol,2004,15(11):2882-2892.
26 Zenker M,Aigner T,Wendler O,et al.Human laminin beta2deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities.Hum Mol Genet,2004,13(21):2625-2632.
27 Goldberg S,Adair-Kirk TL,Senior RM,et al.Maintenance of Glomerular Filtration Barrier Integrity Requires Laminin{alpha}5.J Am Soc Nephrol,2010,21(4):579-586.
28 Duning K,Schurek EM,Schlüter M,et al.KIBRA modulates directional migration of podocytes.JAm Soc Nephrol,2008,19(10):1891-1903.
29 Huber TB,Hartleben B,Winkelmann K,et al.Loss of podocyte aPKClambda/iota causes polarity defects and nephrotic syndrome.JAm Soc Nephrol,2009,20(4):798-806.
30 Hirose T,Satoh D,Kurihara H,et al.An essential role of the universal polarity protein,aPKClambda,on the maintenance of podocyte slit diaphragms.PLoS One,2009,4(1):e4194.
31 Wei C,Möller CC,Altintas MM,et al.Modification of kidney barrier function by the urokinase receptor.Nat Med,2008,14(1):55-63.
32 Beck LH Jr,Bonegio RG,Lambeau G,et al.M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy.N Engl JMed,2009,361(1):11-21.
33 Nguyen T,Di Giovanni S.NFAT signaling in neural development and axon growth.Int JDev Neurosci,2008,26(2):141-145.
34 Rusnak F,Mertz P.Calcineurin:form and function.Physiol Rev,2000,80(4):1483-1521.
35 Tumlin JA.Expression and function of calcineurin in the mammalian nephron:physiological roles,receptor signaling,and ion transport.Am JKidney Dis,1997,30(6):884-895.
36 Balasubramanian L,Sham JSK,Yip KP.Calcium signaling in vasopressin-induced aquaporin-2trafficking.Pflugers Arch,2008,456(4):747-754.
37 Faul C,Donnelly M,Merscher-Gomez S,et al.The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A.Nat Med,2008,14(9):931-938.
38 刘志红,吴 青,汤 曦,等.膜性肾病患者足细胞钙神经蛋白表达的检测及临床意义.肾脏病与透析肾移植杂志,2010,19(1):3-11.
39 Artavanis-Tsakonas S,Rand MD,Lake RJ.Notch signaling:cell fate control and signal integration in development.Science,1999,284(5415):770-776.
40 Cau E,Blader P.Notch activity in the nervous system:to switch or not switch?Neural Dev,2009,4:36.
41 Niranjan T,Bielesz B,Gruenwald A,et al.The Notch pathway in podocytes plays a role in the development of glomerular disease.Nat Med,2008,14(3):290-298.
42 Mundel P,Kriz W.Structure and function of podocytes:an update.Anat Embryol(Berl),1995,192(5):385-397.
43 Zager RA,Johnson AC,Lund S.Uremia impacts renal inflammatory cytokine gene expression in the setting of experimental acute kidney injury.Am J Physiol Renal Physiol,2009,297(4):F961-970.
44 Neal CR,Crook H,Bell E,et al.Three-dimensional reconstruction of glomeruli by electron microscopy reveals a distinct restrictive urinary subpodocyte space.J Am Soc Nephrol,2005,16(5):1223-1235.
45 Neal CR,Muston PR,Njegovan D,et al.Glomerular filtration into the subpodocyte space is highly restricted under physiological perfusion conditions.Am J Physiol Renal Physiol,2007,293(6):F1787-1798.
46 Salmon AH,Toma I,Sipos A,et al.Evidence for restriction of fluid and solute movement across the glomerular capillary wall by the subpodocyte space.Am J Physiol Renal Physiol,2007,293(6):F1777-1786.
47 Foster RR,Slater SC,Seckley J,et al.Vascular endothelial growth factor-C,a potential paracrine regulator of glomerular permeability,increases glomerular endothelial cell monolayer integrity and intracellular calcium.Am J Pathol,2008,173(4):938-948.
48 Satchell SC,Tasman CH,Singh A,et al.Conditionally immortalized human glomerular endothelial cells expressing fenestrations in response to VEGF.Kidney Int,2006,69(9):1633-1640.
49 Foster RR.The importance of cellular VEGF bioactivity in the development of glomerular disease.Nephron Exp Nephrol,2009,113(1):e8-e15.
50 Müller-Deile J,Worthmann K,Saleem M,et al.The balance of autocrine VEGF-A and VEGF-C determines podocyte survival.Am J Physiol Renal Physiol,2009,297(6):F1656-1667.
51 Foster RR,Satchell SC,Seckley J,et al.VEGF-Cpromotes survival in podocytes.Am J Physiol Renal Physiol,2006,291(1):F196-207.
52 Eremina V,Baelde HJ,Quaggin SE.Role of the VEGF—a signaling pathway in the glomerulus:evidence for crosstalk between components of the glomerular filtration barrier.Nephron Physiol,2007,106(2):p32-37.
53 Hohenstein B,Hausknecht B,Boehmer K,et al.Local VEGF activity but not VEGF expression istightly regulated during diabetic nephropathy in man.Kidney Int,2006,69(9):1654-1661.
54 Baelde HJ,Eikmans M,Lappin DW,et al.Reduction of VEGF-A and CTGF expression in diabetic nephropathy is associated with podocyte loss.Kidney Int,2007,71(7):637-645.
55 Levine RJ,Maynard SE,Qian C,et al.Circulating angiogenic factors and the risk of preeclampsia.N Engl J Med,2004,350(7):672-683.
56 Eremina V,Sood M,Haigh J,et al.Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases.J Clin Invest,2003,111(5):707-716.
57 Maciel TT,Kempf H,Campos AH.Targeting bone morphogenetic protein signaling on renal and vascular diseases.Curr Opin Nephrol Hypertens,2010,19(1):26-31.
58 De Petris L,Hruska KA,Chiechio S,et al.Bone morphogenetic protein-7 delays podocyte injury due to high glucose.Nephrol Dial Transplant,2007,22(12):3442-3450.
59 Wang S,de Caestecker M,Kopp J,et al.Renal bone morphogenetic protein-7 protects against diabetic nephropathy.J Am Soc Nephrol,2006,17(9):2504-2512.
60 Collino F,Bussolati B,Gerbaudo E,et al.Preeclamptic sera induce nephrin shedding from podocytes through endothelin-1 release by endothelial glomerular cells.Am J Physiol Renal Physiol,2008,294(5):F1185-1194.
61 Ortmann J,Amann K,Brandes RP,et al.Role of podocytes for reversal of glomerulosclerosisand proteinuria in the aging kidney after endothelin inhibition.Hypertension,2004,44(6):974-981.
62 Barton M.Reversal of proteinuric renal disease and the emerging role of endothelin.Nat Clin Pract Nephrol,2008,4(9):490-501.
63 Dai C,Stolz DB,Bastacky SI,et al.Essential role of integrin-linked kinase in podocyte biology:Bridging the integrin and slit diaphragm signaling.JAm Soc Nephrol,2006,17(8):2164-2175.
64 Yang Y,Guo L,Blattner SM,et al.Formation and phosphorylation of the PINCH-1-integrin linked kinase-alpha-parvin complex are important for regulation of renal glomerular podocyte adhesion,architecture,and survival.J Am Soc Nephrol,2005,16(7):1966-1976.
65 Nagata M,Horita S,Shu Y,et al.Phenotypic characteristics and cyclin-dependent kinase inhibitors repression in hyperplastic epithelial pathology in idiopathic focal segmental glomerulosclerosis.Lab Invest,2000,80(6):869-880.
66 Yamaguchi Y,Iwano M,Suzuki D,et al.Epithelial-mesenchymal transition as a potential explanation for podocyte depletion in diabetic nephropathy.Am JKidney Dis,2009,54(4):653-664.
67 Kang YS,Li Y,Dai C,et al.Inhibition of integrin-linked kinase blocks podocyte epithelial-mesenchymal transition and ameliorates proteinuria.Kidney Int,2010.
68 Jarad G,Cunningham J,Shaw AS,et al.Proteinuria precedes podocyte abnormalities inLamb2-/-mice,implicating the glomerular basement membrane as an albumin barrier.J Clin Invest,2006,116(8):2272-2279.
69 Rastaldi MP,Armelloni S,Berra S,et al.Glomerular podocytes contain neuron-like functional synaptic vesicles.FASEB J,2006,20(7):976-978.
70 Giardino L,Armelloni S,Corbelli A,et al.Podocyte glutamatergic signaling contributes to the function of the glomerular filtration barrier.J Am Soc Nephrol,2009,20(9):1929-1940.
71 Paverstadt H,Kriz W,Kretzler M.Cell biology of the golmerular podocyte.Physiol Rev,2003,83(1):253-307.
