2-苯并咪唑乙酮肟醚衍生物的合成及其抑菌活性
2010-11-26王海波纪增臣滕信焕
王海波, 姜 林, 纪增臣, 滕信焕
(1. 山东农业大学 a. 化学与材料科学学院; b. 植物保护学院, 山东 泰安 271018)
苯并咪唑衍生物具有广泛的生物活性,如杀菌、消炎、驱虫、抗癌等作用[1,2]。农药中的苯并咪唑类杀菌剂,如多菌灵、苯菌灵和噻菌灵,具有高效、广谱、内吸性强的特点,至今仍活跃在杀菌剂市场。然而,由于使用了40年之久,病害菌的抗性随之增强,导致该类杀菌剂的活性降低[3]。因此,研制新型高效的苯并咪唑类杀菌剂有重要的应用价值。
肟醚衍生物也是一类具有杀虫、除草和杀菌等生物活性的化合物[4,5], 如高效杀菌剂肟菌酯[6]和肟醚菌胺[7],因而在新农药创制中, 肟醚结构常常被选为有效的活性基团。
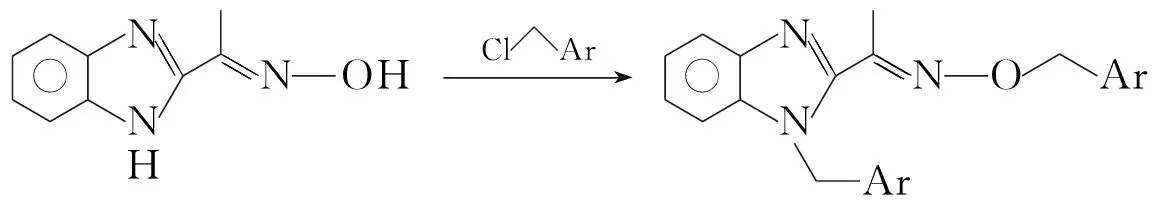
为寻找优良杀菌活性的化合物,本文根据活性结构拼接原理, 将肟醚活性亚结构引入到苯并咪唑母体结构中,设计并合成了6个新型的2-苯并咪唑乙酮烷基肟醚(3a~3c)和1-取代苄基-2-苯并咪唑乙酮苄基肟醚(3d~3f, Scheme 1),并初步测试了3的抑菌活性。
1 实验部分
1.1 仪器与试剂
WRS-1A型数字熔点仪(温度计未校正);美国Varian公司Inova-600型核磁共振仪(CDCl3为溶剂,TMS为内标);美国Nicolet 380型傅立叶变换红外光谱仪(KBr压片);德国Elementar VarioEL Ⅲ型元素分析仪;宁波江南仪器厂GXZ型智能光照培养箱。

23d~3f
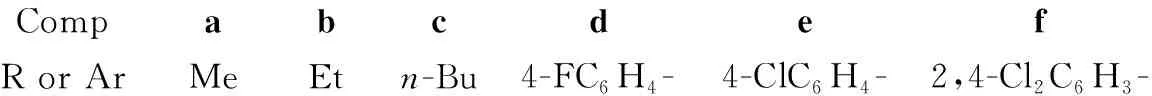
CompabcdefR or ArMeEtn-Bu4-FC6H4-4-ClC6H4-2,4-Cl2C6H3-
Scheme1
2-苯并咪唑乙酮(1)按文献[8]方法制备;其余所用试剂均为市售分析纯或化学纯。
1.2 合成
(1)3a~3c的合成
在三颈瓶中加入1 1.8 g(10 mmol),无水碳酸钠1.3 g(12 mmol),乙醇10 mL,冰浴冷却,搅拌下滴加烷氧基胺盐酸盐(10 mmol)的水溶液5 mL(15 min内),回流反应5 h。蒸去大部分乙醇,残余液倒入10 g碎冰中,抽滤,滤饼经硅胶柱层析[洗脱剂:V(乙酸乙酯) ∶V(石油醚)=1 ∶4] 纯化得白色晶体3a~3c。
(2)3d~3f的合成
在三颈瓶中加入1 4.0 g(25 mmol),无水碳酸钠3.4 g(32 mmol),乙醇25 mL,冰浴冷却,搅拌下滴加盐酸羟胺2.3 g(32 mmol)的水溶液12 mL(10 min内),回流反应5 h。蒸去乙醇,剩余液倒入25 g碎冰中,抽滤,滤饼用混合溶剂[V(丙酮) ∶V(石油醚)=1 ∶2]重结晶得白色固体2-苯并咪唑乙酮肟(2) 4.1 g,产率93.2%, m.p.245 ℃ ~ 247 ℃;1H NMRδ: 2.48(s, 3H, CH3), 7.26~7.82(m, 4H, ArH), 9.63(s, 1H, NH); IRν: 3 434(N-H), 1 597(C=N) cm-1; Anal.calcd for C9H9N3O: C 61.70, H 5.18, N 23.99; found C 61.48, H 5.26, N 23.69。
在两颈瓶中加入20.88 g(5 mmol),氢氧化钠0.32 g(8 mmol),四丁基碘化铵0.05 g(0.14 mmol),丙酮20 mL,取代苄氯10 mmol,搅拌下于40 ℃~50 ℃反应(TLC跟踪)。蒸去丙酮,残余物中加入二氯甲烷,有机层经水洗,无水硫酸镁干燥,脱溶剂得淡黄色固体,用乙醇重结晶得白色晶体3d~3f。
3a: 产率67.7%, m.p.202 ℃~203 ℃;1H NMRδ: 2.41(s, 2.64H, CH3,E-isomer), 2.51(s, 0.36H, CH3,Z-isomer), 4.02(s, 2.63H, OCH3,E-isomer), 4.13(s, 0.37H, OCH3,Z-isomer), 7.26~7.87(m, 4H, ArH), 9.99(s, 1H, NH); IRν: 3 444(N-H), 1 619(C=N) , 1 051(=N-O-C) cm-1; Anal.calcd for C10H11N3O: C 63.48, H 5.86, N 22.21; found C 63.84, H 5.84, N 22.12。
3b: 产率64.5%, m.p.149 ℃ ~ 151 ℃;1H NMRδ: 1.36(t,J=7.2 Hz, 2.61H, CH2CH3,E-isomer), 1.45(t,J=7.2 Hz, 0.39H, CH2CH3,Z-isomer), 2.42(s, 2.61H, CH3,E-isomer), 2.51(s, 0.39H, CH3,Z-isomer), 4.28(q,J=7.2 Hz, 1.75H, OCH2,E-isomer), 4.39(q,J=7.2 Hz, 0.25H, OCH2,Z-isomer), 7.24~7.81(m, 4H, ArH), 9.84(s, 1H, NH); IRδ: 3 448(N-H), 1 623(C=N), 1 052(=N-O-C) cm-1; Anal.calcd for C11H13N3O: C 65.01, H 6.45, N 20.68; found C 65.22, H 6.42, N 20.73。
3c: 产率67.8%, m.p.126 ℃~127 ℃;1H NMRδ: 0.96 ~ 1.01(m, 3H, CH2CH3,Z+E-isomers), 1.41~1.49(m, 2H, CH2CH3,Z+E-isomers), 1.68~1.74(m, 1.55H, OCH2CH2,E-isomer), 1.79~1.85(m, 0.45H, OCH2CH2,Z-isomer), 2.41(s, 2.27H, CH3,E-isomer), 2.51(s, 0.73H, CH3,Z-isomer), 4.23(t,J=7.2 Hz, 1.51H, OCH2,E-isomer), 4.34(t,J=7.2 Hz, 0.49H, OCH2,Z-isomer), 7.24~7.88(m, 4H, ArH), 9.84(s, 1H, NH); IRν: 3 444(N-H), 1 623(C=N), 1 066(=N-O-C) cm-1; Anal.calcd for C13H17N3O: C 67.51, H 7.41, N 18.17; found C 67.96, H 7.41, N 18.23。
3d: 产率67.7%, m.p. 104 ℃ ~ 106 ℃;1H NMRδ: 2.51(s, 3H, CH3), 5.05(s, 2H, OCH2), 5.59(s, 2H, NCH2), 6.92~7.82(m, 12H, ArH); IRν: 1 606 (C=N), 1 010 (=N-O-C) cm-1; Anal.calcd for C23H19N3OF2: C 70.58, H 4.89, N 10.74; found C 70.75, H 5.01, N 10.39。
3e: 产率72.6%, m.p. 130 ℃ ~ 132 ℃;1H NMRδ: 2.51(s, 3H, CH3), 5.04(s, 2H, OCH2), 5.57(s, 2H, NCH2), 6.84~7.29(m, 12H, ArH); IRν: 1 590(C=N), 1 010(=N-O-C) cm-1; Anal.calcd for C23H19N3OCl2: C 65.10, H 4.51, N 9.90; found C 65.57, H 4.79, N 9.45。
3f: 产率62.6%, m.p. 158 ℃ ~ 160 ℃;1H NMRδ: 2.50(s, 3H, CH3), 5.12(s, 2H, OCH2), 5.68(s, 2H, NCH2), 6.08~7.80(m, 10H, ArH); IRν: 1 617(C=N), 1 018(=N-O-C) cm-1; Anal.calcd for C23H17N3OCl4: C 56.01, H 3.47, N 8.52; found C 55.98, H 3.75, N 8.33。
2 结果与讨论
2.1 合成方法
合成3设计了两条路线:烷氧基肟醚(3a~3c)采用1与烷氧基胺盐酸盐在碱性条件下直接反应生成;而苄氧基肟醚(3d~3f) 采取1与盐酸羟胺反应先制取酮肟(2); 2再与取代苄氯作用得到产物。
实验发现,合成3a~3c时,若用2与卤代烷反应的策略,由于碘甲烷、溴乙烷沸点较低以及正溴丁烷活性相对较低等原因,产率很低(不超过40%)。采用Scheme 1方法,以乙醇水溶液作反应介质,路线简洁、产物直接由介质中析出、产率较高。合成3d~3f时,由于取代苄氧基胺盐酸盐制备成本高,因而本文以价格相对低廉的盐酸羟胺和取代苄氯为原料,经两步反应的方法制得产物。但是,由于取代苄氯反应活性较高,最后得到是N1-H也被苄基取代的肟醚。
2.2 3的几何异构体
由于C=N双键的存在,肟醚类化合物有时会出现顺反异构现象[9,10]。从1H NMR谱图上可看出,3a~3c均出现了Z式和E式异构体,通过计算1H NMR谱图上两个异构体同一质子相应吸收峰积分面积,可以推知Z式和E式的比例在1.0 ∶3.5~1.0 ∶7.3,即E式所占比例大,其原因可能是E式结构中与醚键相连的烷基与苯并咪唑环处在相反方向,空间位阻较小的缘故。3d~3f由于取代苄基体积较大,只得到稳定性较高的E式异构体。
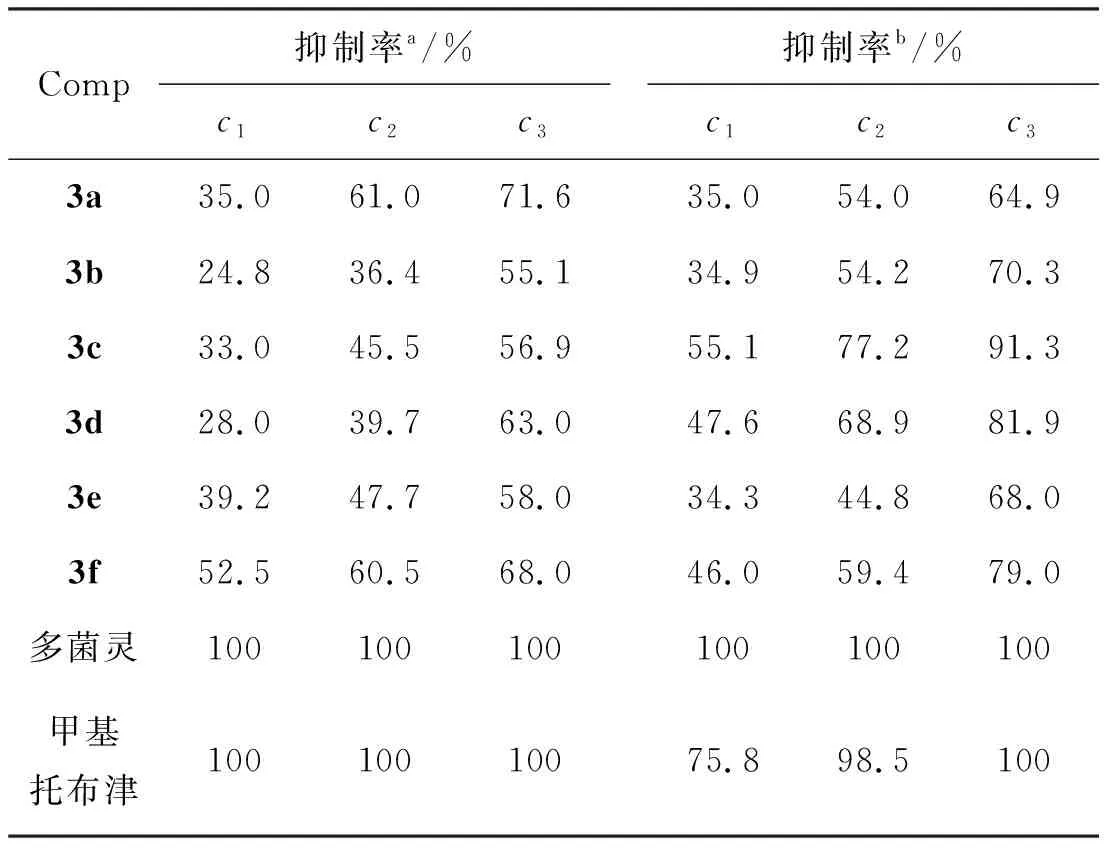
表 1 3的抑菌活性*Table 1 Fungicidal activity of 3
*c1=20 mg · L-1,c2=40 mg · L-1,c3=80 mg · L-1;a对番茄灰霉病菌;b对黄瓜菌核病菌
2.3 3的抑菌活性
采用菌丝生长速率法[11]测定目标化合物的抑菌活性。以番茄灰霉病菌(Botrytiscinereapers)和黄瓜菌核病菌(Sclerotiniasclerotiorum)为测试菌种,以苯并咪唑类杀菌剂多菌灵、甲基托布津为对比药剂。
按培养基与药液体积比9 ∶1的比例制成含药培养基(马铃薯-葡萄糖-琼胶,PDA),微波炉溶解后制成浓度分别为20 mg · L-1, 40 mg · L-1, 80 mg · L-1的含药平板,以加入等体积无菌水的培养基平板为对照。然后将直径4 mm的菌饼反接到含药平板中央,置于(25±1) ℃下培养96 h,每处理设3个重复。用十字交叉法测量菌落直径,每个菌落测量2次,以其平均值代表菌落的大小,按下式计算抑制率,测试结果见表1。由表1可知,3对黄瓜菌核病菌有一定的抑制活性,c(3)=80 mg · L-1时抑制率在58.0%~71.6%;而对番茄灰霉病菌有较高的抑制活性,其中以3c活性最高,抑制率达到91.3%。与商品化的该类杀菌剂多菌灵和甲基托布津相比,其抑菌活性还有差距。
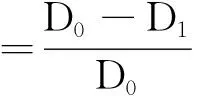
式中:D0为对照组菌落直径;D1为处理组菌落直径
[1] Madkour H M F, Farag A A, Ramses S S,etal. Synthesis and fungicidal activity of new imidazoles from 2-(chloromethyl)-1H-benzimidazole[J].Phosphorus,Sulfur,and Silicon,2006,181:255-265.
[2] 李焱,马会强,王玉炉. 苯并咪唑及其衍生物合成与应用研究进展[J].有机化学,2008,28:210-217.
[3] 崔文辉,张有明,魏太保. 2-芳氧甲基苯并咪唑类化合物的合成[J].合成化学,2006,14:364-367.
[4] 范磊,崔建国,韦英亮,等. 具有生物活性的肟醚类化合物的研究进展[J].现代农药,2008,7(4):6-10.
[5] 宋宝安,刘新华,杨松,等. 肟类衍生物的合成与农药生物活性的研究进展[J].有机化学,2005,25:507-525.
[6] Margot P, Huggenberger F, Amrein J,etal. CGA 279202:A new broad-spectrum strobilourin fungicide[C].The British Crop Protection Council Conference Pest & Diseases 1998:Proceedings of an International Conference,UK:Brighton Hilton Metropole Hotel,1998:375-382.
[7] Thomas G S, Herbert B M, Ruth M F,etal. Phenylacetic acid derivatives,processes and intermediates for use in producing them and agents containing them[P].US: 5 948 932,1997.
[8] Cheeseman G W H. 2-Acetylbenzimidazole[J].J Chem Soc,1964:4645-4646.
[9] 江黎黎,陈超南,周延菲,等. 含肟醚的新型三唑并嘧啶衍生物的合成及除草活性研究[J].有机化学,2009,29:1392-1404.
[10] 刘天麟,周懿波. 哒嗪酮-类肟醚菊酯衍生物的合成研究[J].有机化学,2000,20:758-763.
[11] 慕立义. 植物化学保护研究法[M].北京:中国农业出版社,1994.
