2-氰基-3,4-二氢喹唑啉衍生物的合成
2010-11-26陈金萍朱丽娜周海波付德才徐柏玲
陈金萍, 朱丽娜, 周海波, 付德才, 徐柏玲
(1. 中国医学科学院 北京协和医学院 药物研究所,北京 100050; 2. 河北科技大学
3,4-二氢喹唑啉是含有两个氮原子的一类重要的苯并杂环化合物,具有多种生物活性[1~3]。 Tpl2(Tumor progression locus 2)是调控促炎症细胞因子TNF-α的产生及信号传导的重要激酶之一,Tpl2激酶抑制剂有望成为新型的抗炎药物[4]。研究表明,3-氰基喹啉类化合物是高效的Tpl2激酶抑制剂[5]。基于已有的构效关系,本课题组设计了2-氰基-3,4-二氢喹唑啉类化合物,以期寻找新型的Tpl2激酶抑制剂。
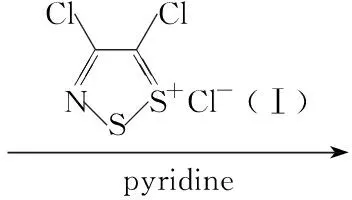
尽管合成3,4-二氢喹唑啉类衍生物有许多方法[6~9],但是2-氰基-3,4-二氢喹唑啉的合成还未见文献报道。本文将邻硝基苯甲醛与取代芳胺(1a~1e)或取代苄胺(2f~2j)进行还原氨化反应,得到N-(2-硝基苄基)取代苯胺(3a~3e)或N-(2-硝基苄基)取代苄胺(4f~4j);将3或4的2-位硝基还原为氨基,得到关键中间体N-(2-氨基苄基)取代苯胺(5a~5e)或N-(2-氨基苄基)取代苄胺(6f~6j); 5或6分别与4,5-二氯-1,2,3-二噻唑氯化物(Ⅰ)[10](作为氰基的合成子,曾被广泛地用于合成氰基取代的杂环化合物,如:苯噁唑[11]、苯并噻唑[12,13]、苯并噁嗪[14]、咪唑[15]和喹唑啉类[16~19])反应生成N-芳基亚胺基-1,2,3-二噻唑(Ⅱ或Ⅲ),在不需要分离的情况下,Ⅱ或Ⅲ中的二级胺氮原子作为亲核体,直接进攻二噻唑环的5-C位,发生分子内加成消除反应,合成了一系列2-氰基-3,4-二氢喹唑啉衍生物(7a~7e和8f~8j)(Scheme 1),其中3c~3e,4h~4j,5a,5c~5e,6h~6j,7a~7d,8f~8j未见文献报道,其结构经1H NMR和HR-MS表征。
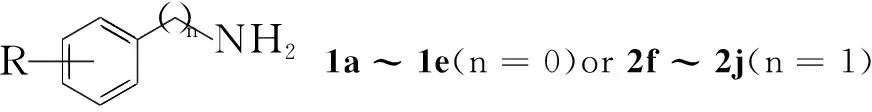
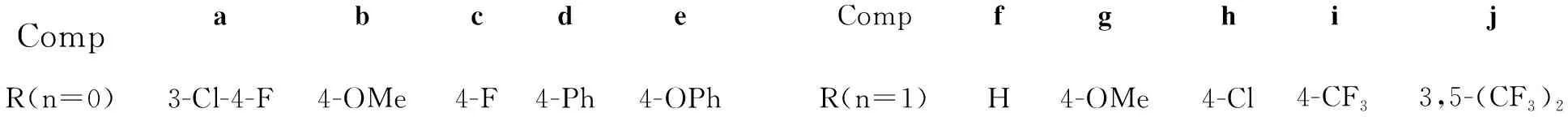
CompabcdeCompfghijR(n=0)3-Cl-4-F4-OMe4-F4-Ph4-OPhR(n=1)H4-OMe4-Cl4-CF33,5-(CF3)2
Scheme1
1 实验部分
1.1 仪器与试剂
500D型熔点仪;Mercury 300型核磁共振仪(CDCl3为溶剂,TMS为内标);LC/MSDTOF型质谱仪;CEM公司Explorer 24型微波合成仪。
硅胶200目~300目,青岛海洋化工厂;其余所用试剂均为市售分析纯。
1.2 中间体的合成
(1) 3和4的合成(以3a和4f为例)
①3a的合成
将邻硝基苯甲醛151 mg(1 mmol)和3-氯-4-氟苯胺(1a)145 mg(1 mmol)溶于异丙醇(1 mL)中,置微波反应器中,在功率150 W,压力150 PSI的条件下,于130 ℃辐射3 min。蒸除异丙醇,残余物用甲醇(5 mL)溶解;加入硼氰化钠38 mg(1 mmol),于室温搅拌反应15 min。蒸除甲醇,用二氯甲烷(40 mL)溶解后依次用蒸馏水(10 mL),饱和碳酸氢钠溶液(10 mL)和饱和氯化钠溶液(10 mL)洗涤,无水硫酸钠干燥,浓缩后经硅胶柱层析[洗脱剂:A=V(石油醚) ∶V(乙酸乙酯)=6 ∶1]纯化得黄色固体3a。用类似方法合成3b~3e。
②4f的合成
在反应瓶中加入邻硝基苯甲醛604 mg(4 mmol)的甲醇(10 mL)溶液,搅拌下加入苄胺(2f)428 mg(4 mmol),于室温反应1 h。加入硼氰化钠142 mg(4 mmol)和催化量的醋酸(5滴),继续反应30 min。加水淬灭反应,浓缩后加入二氯甲烷50 mL,依次用蒸馏水(10 mL),饱和碳酸氢钠溶液(10 mL)和饱和氯化钠溶液(10 mL)洗涤,无水硫酸钠干燥,浓缩后经硅胶柱层析(洗脱剂:A=3 ∶1)分离得橙色油状物4f。用类似方法合成4g。
③4j的合成
在反应瓶中加入邻硝基苯甲醛453 mg(3 mmol)的二氯甲烷(10 mL)溶液,搅拌下加入3,5-双三氟甲基苄胺(2j)2.19 g(9 mmol),三乙酰氧基硼氰化钠1.9 g(9 mmol)及催化量醋酸(5滴),于室温反应23 h(TLC检测)。加入二氯甲烷50 mL,依次用蒸馏水(10 mL),饱和碳酸氢钠溶液(10 mL)和饱和氯化钠溶液(10 mL)洗涤,无水硫酸钠干燥,浓缩后经硅胶柱层析(洗脱剂:A=6 ∶1)分离得白色固体4j。用类似方法合成4h和4i。
(2) 5和6的合成(以5a和6j为例)
①5a的合成
在反应瓶中加入3a228 mg(0.81 mmol)的醋酸(5 mL)溶液,搅拌下加入锌粉318 mg(4.88 mmol),于室温反应55 min。过滤,浓缩滤液,加入二氯甲烷50 mL,依次用蒸馏水(10 mL),饱和碳酸氢钠溶液(10 mL)和饱和氯化钠溶液(10 mL)洗涤,无水硫酸钠干燥,浓缩后经硅胶柱层析(洗脱剂:A=4 ∶1)分离得黄色固体5a。用类似方法合成5b~5e。
②6j的合成
在反应瓶中加入4j300 mg(0.79 mmol)的乙醇(5 mL)溶液,搅拌下加入氯化亚锡754 mg(3.97 mmol)和浓盐酸474 mg(4.74 mmol),回流反应10 min。用10%氢氧化钠溶液调至pH 10,用二氯甲烷(3×40 mL)萃取,无水硫酸钠干燥,浓缩后经硅胶柱层析(洗脱剂:A=10 ∶1)分离得淡黄色油状物6j。用类似方法合成6f~6i。
1.3 目标化合物7和8的合成(以7d为例)
在反应瓶中加入5d125 mg(0.46 mmol)的二氯甲烷(8 mL)溶液,搅拌下加入Ⅰ 133 mg(0.64 mmol),缓慢滴加吡啶72 mg(0.91 mmol)的二氯甲烷(2 mL)溶液,于室温反应4 h。加入二氯甲烷50 mL,依次用饱和硫酸铜溶液(10 mL)和饱和氯化铵溶液(10 mL)洗涤,无水硫酸钠干燥,浓缩后经硅胶柱层析(洗脱剂:A=18 ∶1)纯化得黄色固体7d。用类似的方法合成7a~7c,7e,8f~8j。
2 结果与讨论
中间体和目标化合物的实验数据见表1,中间体的光谱数据见表2,目标化合物的光谱数据见表3。
2.1 合成
(1) 3和4的合成
邻硝基苯甲醛与1或2进行还原氨化反应,生成3或4。依据1或2结构的不同,采用了不同的反应条件。当2f或2g与邻硝基苯甲醛进行还原氨化反应时,以醋酸为催化剂,硼氢化钠为还原剂,4f和4g的收率分别为74.2%和83.3%(表1)。在同样的反应条件下,当苄胺的苯环上取代基为Cl(2h), CF3(2i)和3,5-(CF3)2(2j)时,未能得到预期产物4h~4j,而是分离得到了邻硝基苯甲醛的还原产物邻硝基苯甲醇。这是由于2h~2j的亲核性降低,席夫碱的生成速度小于硼氢化钠还原邻硝基苯甲醛的速度。但当采用还原能力相对较弱的三乙酰氧基硼氢化钠为还原剂时,4h~4j的收率大于80%(表1)。
当反应底物为芳胺(1)时,以三乙酰氧基硼氢化钠为还原剂,1与邻硝基苯甲醛反应,不仅反应时间长并且有原料剩余。为此本文采用了微波辐射方法,在不需要任何催化剂的条件下,1a~1e与邻硝基苯甲醛反应,生成席夫碱;然后以硼氢化钠为还原剂,得到3a~3e(表1)。该方法不仅反应时间短,而且收率高。
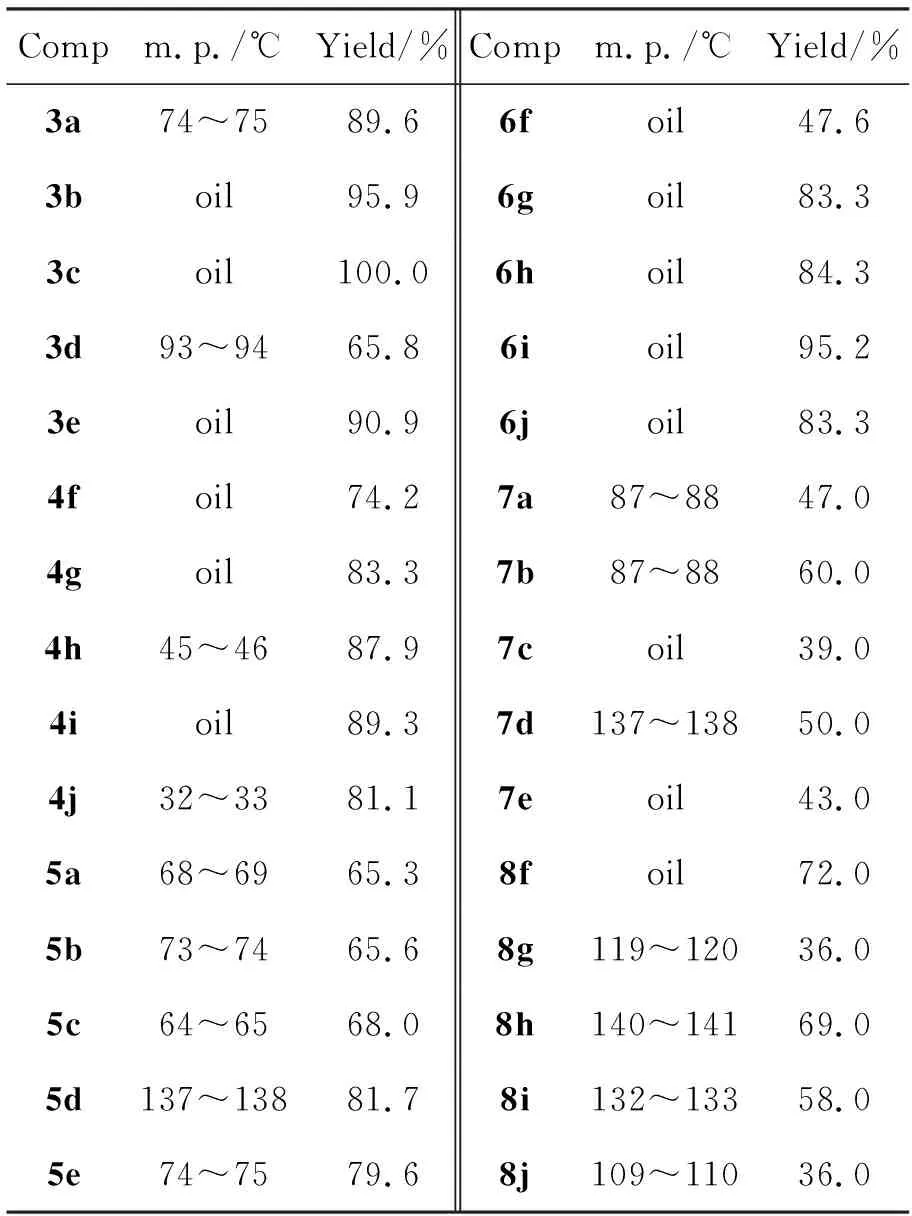
表1 化合物的实验数据Table 1 Experimental data of compounds
(2) 5和6的合成
当反应底物为4f~4j时,采用锌粉-醋酸体系还原硝基,未能分离得到产物;采用氯化亚锡-浓盐酸作为还原剂,除6f外,6g~6j的收率均大于80%。当反应底物3a~3e时,采用锌粉-醋酸体系进行还原反应,则可以顺利得到5a~5e,收率65.3%~81.7%(表1)。
(3) 7和8的合成
5或6与Ⅰ反应,生成中间体Ⅱ或Ⅲ,通过分子内亲核加成消除反应,生成环合产物7或8,收率36.0%~72.0%(表1)。由表1可以看出,8f~8j的收率略高于7a~7e,这是由于Ⅱ中N原子的亲核性低于Ⅲ的缘故。同样,在8f~8j系列化合物中,随着取代基拉电子能力的增加(8f,8h~8j),化合物的收率下降。值得注意的是,当取代基为甲氧基时,由于中间体Ⅱg的收率较低,导致8g的收率低于8f。当反应底物为5a~5e,供电子基团如甲氧基有利于环合7b的生成,收率为60.0%,高于吸电子基团取代的7a和7c~7e。

表2 中间体的1H NMR数据Table 2 1H NMR data of intermediates
续表2

Comp1H NMR δ(J/Hz)6g7.24(m, 2H), 7.09(td, J=7.8, 1.8, 1H), 7.02(m, 1H), 6.84~6.89(dt, J=8.7, 2.1, 2H), 6.64~6.72(td, J=7.5, 0.9, 2H), 3.81(s, 5H), 3.73(s, 2H)6h7.28(d, J=8.1, 2H), 7.23(d, J=8.7, 2H), 7.08(t, J=7.5, 1H), 7.00(d, J=7.5, 1H), 6.68(t, J=8.1, 2H), 3.80(s, 2H), 3.74(s, 2H)6i7.58(d, J=8.1, 2H), 7.43(d, J=8.1, 2H), 7.12(t, J=7.5, 1H), 7.04(d, J=7.2, 1H), 6.69(t, J=7.5, 2H), 3.85(s, 2H), 3.84(s, 2H)6j7.76(s, 3H), 7.10(t, J=7.5, 1H), 7.04(d, J=7.5, 1H), 6.69(d, J=7.5, 1H), 6.65(d, J=7.5, 1H), 4.45(brs, 2H), 3.90(s, 2H), 3.86(s, 2H)
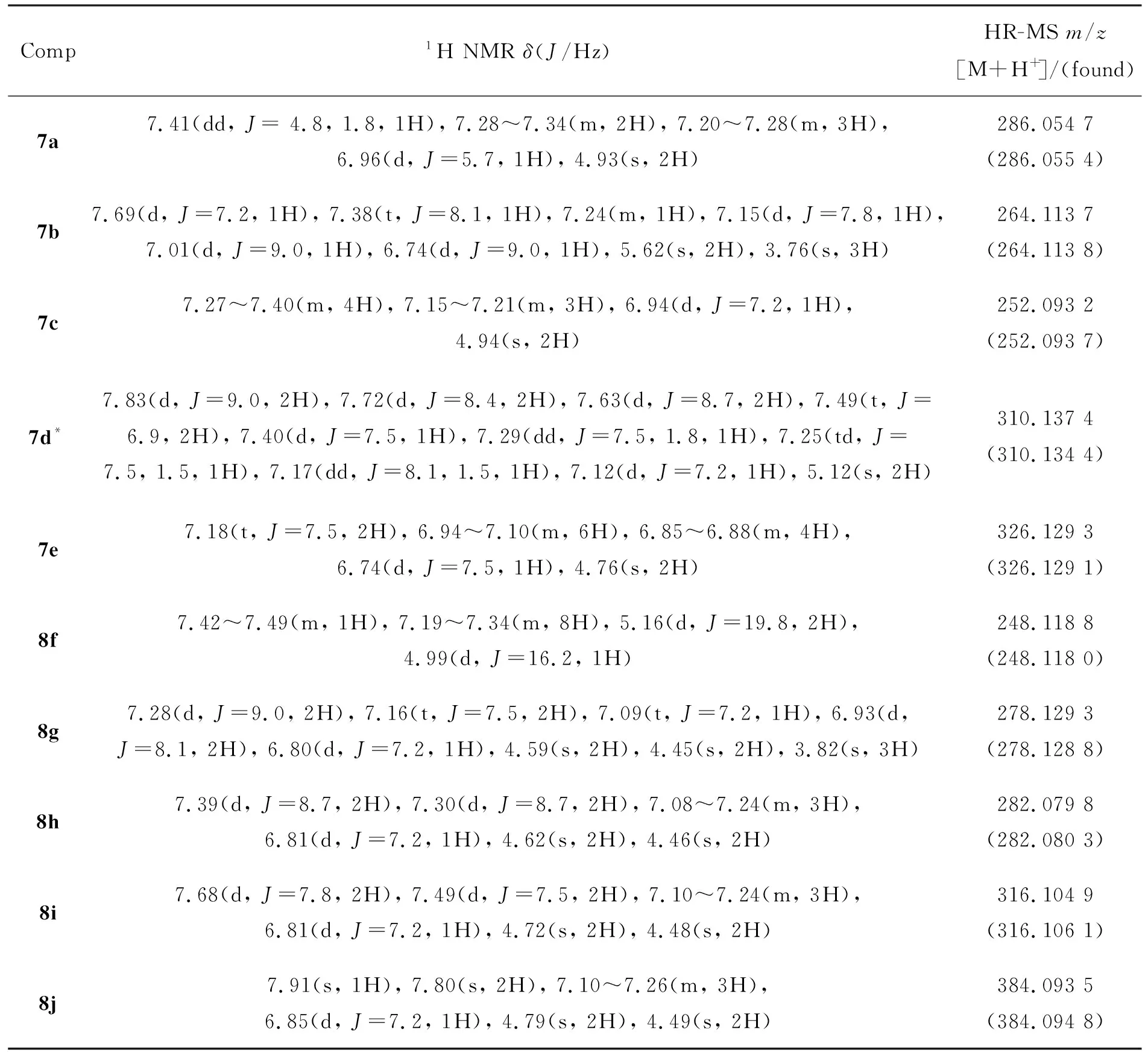
表3 目标化合物的光谱数据Table 3 Spectra data of target compounds
*氘代丙酮为溶剂
3 结论
采用4,5-二氯-1,2,3-二噻唑氯化物作为氰基的合成子,建立了一种新的2-氰基-3,4-二氢喹唑啉合成方法,为本课题组寻找2-氰基-3,4-二氢喹唑啉类Tpl2激酶抑制剂奠定了基础。
[1] Ishikawa F, Watanabe Y, Saegusa J. Cycli guanidines.Ⅸ.Synthesis of 2-amino-3,4-dihydroquinazolines as blood platelet aggregation inhibitors[J].Chem Pharm Bull,1980,28:1357-1364.
[2] Baxter E W, Conway K A, Kennis L,etal. 2-Amino-3,4-dihydroquinazolines as inhibitors of BACE-1(β-Site APP Cleaving Enzyme):Use of structure based design to convert a micromolar hit into a nanomolar lead[J].J Med Chem,2007,50:4261-4264.
[3] Seo H N, Choi J Y, Choe Y J,etal. Discovery of potent T-type calcium channel blocker[J].Bioorg Med Chem Lett,2007,17:5740-5743.
[4] Dumitru C D, Ceci J D, Tsatsanis C,etal. TNF-αinduction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway[J].Cell,2000,103:1071-1083.
[5] Green N, Hu Y, Janz K,etal. Inhibitors of tumor progression loci-2(Tpl2) kinase and tumor necrosis factorα(TNF-α) production:Selectivity and in vivo antiinflammatory activity of novel 8-substituted-4-anilino-6-aminoquinoline-3-carbonitriles[J].J Med Chem,2007,50:4728-4745.
[6] Zhang J F, B J L, Lou B L,etal. Solid-phase synthesis of 3,4-dihydroquinazoline derivatives[J].Tetrahedron Lett,2001,42:8405-8408.
[7] Wang F J, Hauske J R. Solid-phase synthesis of 3,4-dihydroquinazoline[J].Tetrahedron Lett,1997,38:8651-8654.
[8] Schmidt R R. Synthese von 3,4-dihydrochinazolinen durch polar 1,4-cycloaddition[J].Tetrahedron Lett,1968,9:3443-3446.
[9] Lee B H, Lee J Y, Chung B Y,etal. Synthesis of 2-substituted 3,4-dihydroquinazoline derivatives via regioselective addition of a carbon nucleophile to a carbodiimide[J].Heterocycles,2004,63:95-105.
[10] Appel R, Janssen H, Siray M,etal. Synthese und reaktionen des 4,5-dichlor-1,2,3-dithbiazoliumchlorids[J].Chem Ber,1985,118:1632-1643.
[11] Rees C W. Polysulfur-nitrogen heterocyclic chemistry[J].J Heterocycl Chem,1992,29:639-651.
[12] Bénéteau V, Besson T, Rees C W. Rapid synthesis of 2-cyanobenzothiazoles fromN-aryliminodithiazoles under microwave irradiation[J].Synth Commun,1997,27:2275.
[13] English R F, Rakitin O A, Rees C W,etal. Conversion of imino-1,2,3-dithiazoles into 2-cyanobenzothiazoles,cyanoimidoyl chlorides and diatomic sulfur[J].J Chem Soc,Perkin Trans 1,1997:201-206.
[14] Besson T, Emayan K, Rees C W. 3,1-Benzoxazin-4-ones,3,1-benzothiazin-4-ones andN-arylcyanothioformamides[J].J Chem Soc,Chem Commun,1995:1419-1420.
[15] Konstantinova L S, Rakitin O A, Rees C W,etal. New route to 2-cyanobenzimidazoles[J].Tetrahedron,1998,54:9639-9650.
[16] Besson T, Dozias M J, Guillard J,etal. Expeditious routes to 4-alkoxyquinazoline-2-carbonitriles and thiocarbamates viaN-arylimino-1,2,3-dithiazoles using microwave irradiation[J].Tetrahedron,1998,54:6475-6484.
[17] Lee H S, Chang Y G, Kim K, A facile synthesis of 3-substituted 2-syanoquinazolin-4(3H)-ones and 3-slkyl-2-cyanothieno[3,2-d]pyrimidin-4(3H)-ones via 1,2,3-dithiazoles[J].J Heterocyclic Chem,1998,35:659-668.
[18] Chang Y G, Kim K. Synthesis of 3-aryl-3,4-dihydro-4-hydroxy-4-phenylquinazoline-2-carbonitrile via 2-(benzoyl)arylimino-4-chloro-5H-1,2,3-dithiazoles[J].Synlett,2002:1423-1426.
[19] Jeon M K, Kim D S, La H J,etal. Solid-phase synthesis of 2-cyanoquinazolin-4(3H)-one and 2,3-dihydrooxazolo[2,3-b]quinazolin-5-one derivatives utilizing resin-bound anthranilic acid derivatives[J].Tetrahedron Lett,2005,46:7477-7481.
