新型5H-茚[1,2-b]吡啶和11H-茚[1,2-b]喹啉三氟甲磺酸盐衍生物的合成其及抗肿瘤活性*
2010-11-26闻娣娣刘启伦李秋莲杭臣臣朱永明
闻娣娣, 刘启伦, 李秋莲, 杭臣臣, 朱永明
(苏州大学 药学院,江苏 苏州 215123)
由于抗肿瘤药物往往在杀死肿瘤细胞的同时对正常组织细胞也有较强的毒副作用,因此寻找低毒、高效的抗肿瘤药物成了人类新药研发的不懈目标。Hajime Katayama课题组[1,2]报道了3-氧-3H-吡唑并[1,5-a]吲哚类化合物对癌细胞L1210, Hela, PC-1, KAT0Ⅲ, LoVo及MCF-7的细胞毒性以及对鼠白血病细胞L1210的抗肿瘤活性。进一步研究发现一些4H-吡唑并[1,5-a]吲哚类衍生物的三氟甲磺酸盐衍生物也具有很强的抑制DNA拓扑异构酶Ⅰ和Ⅱ活性。
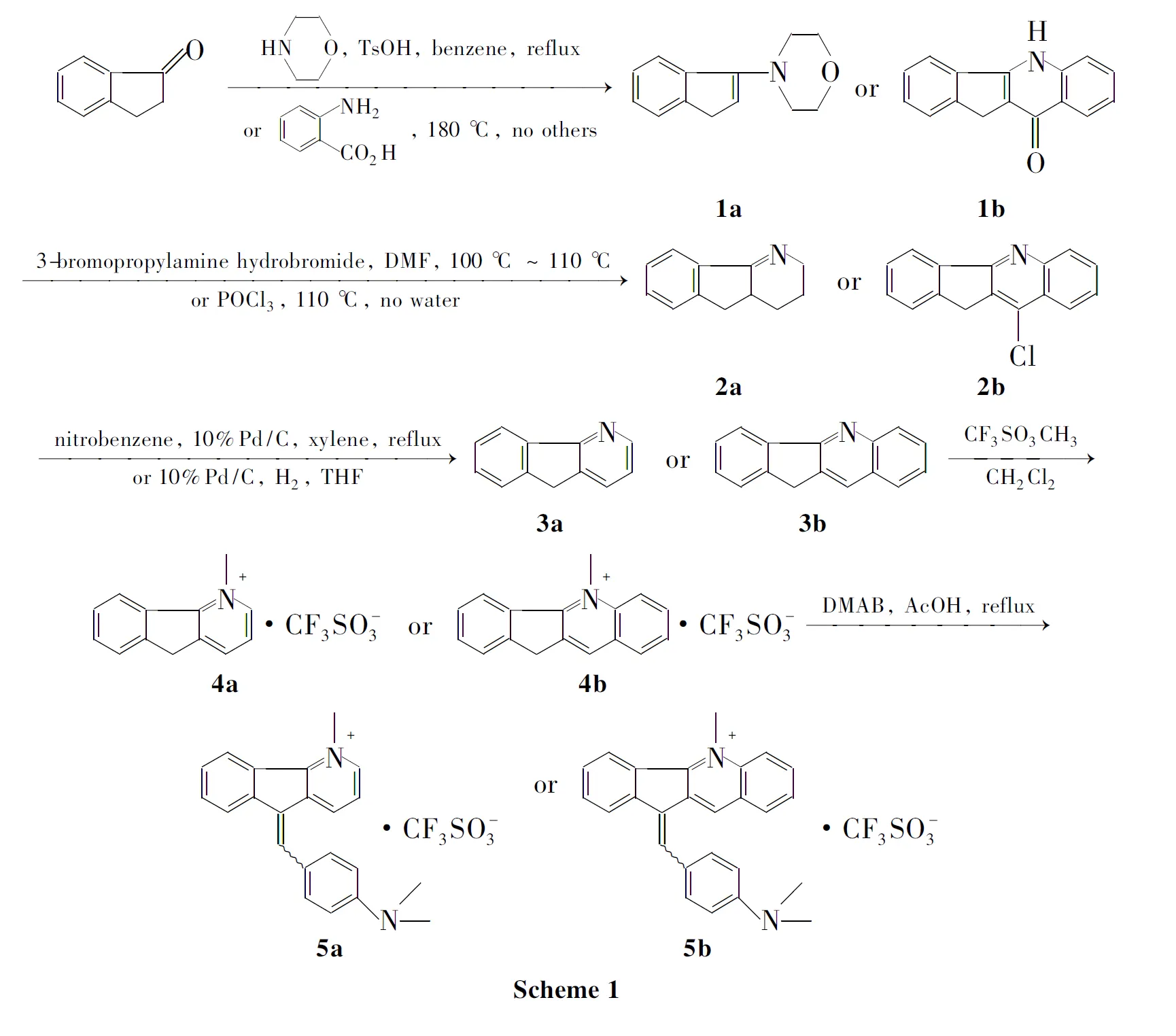
本课题组通过设计新的合成路线,在吡唑并[1,5-a]吲哚的母核(A, Chart 1)上引入多种官能团[3],对K562和HL60肿瘤细胞的毒性试验和构效关系的研究表明此类化合物有很好的体外抗肿瘤活性。通过构效关系分析和生物电子等排设计药物的原理,由A我们设想到:将吲哚环的N原子用C原子替换,吡唑环变为吡啶环,吡啶替代吡唑得到的新化合物可能具有更高的生物活性、更低的毒性或更高的生物利用度,因此,本文以1-茚酮为原料,设计并合成了两个新型结构的先导化合物:1-甲基-5-(4-二甲氨基苄烯)-5H-茚[1,2-b]吡啶三氟甲磺酸盐(5a)和5-甲基-11-(4-二甲氨基苄烯)-11H-茚[1,2-b]喹啉三氟甲磺酸盐(5b)(Scheme 1),其结构经NMR, IR和MS表征。采用MTT法,以HEP-2(人喉鳞癌细胞)和K562细胞(人红白血病细胞)为测试肿瘤细胞株对5a和5b进行了体外抗肿瘤活性检测。
A
Chart1
1 实验部分
1.1 仪器与试剂
XT-5型显微熔点仪(温度未校正);美国瓦利安公司400 MHz型核磁超导共振谱仪(CDCl3为溶剂,TMS为内标);美国瓦利安公司1000 FT-IR型红外光谱仪(KBr压片);英国Micromass公司GCT-TOF型高分辨质谱仪;美国BIO-RAD伯乐公司80型全自动酶标仪。
RPMl l640培养基,GIBCO公司;HEP-2, K562肿瘤细胞株及四氮哇盐(MTT),Sigma公司;其余所用试剂均为分析纯。
1.2 中间体的合成
(1) 1的合成
将1-茚酮5.28 g(40 mmol)溶于苯(25 mL)中,搅拌下加入吗啡啉9 mL(100 mmol ),催化剂量的甲苯-4-磺酸,回流分水至油水分离器不再有水生成。减压蒸馏,残余物为琥珀色油状液体1a3.02 g,收率37.56%, m.p.41.5 ℃~42.5 ℃。
将1-茚酮0.46 g(3.5 mmol)与邻氨基苯甲酸0.28 g(2 mmol)加热熔融,搅拌下回流(180 ℃)反应1.5 h。反应物用吡啶和乙醚洗涤,干燥得黄色固体1b0.32 g,收率67.6%, m.p.360 ℃~365 ℃。
(2) 2的合成
将3-溴丙胺氢溴酸盐2.19 g(10 mmol)溶于干燥的DMF(10 mL)中,搅拌下加入1a2.21 g(11 mmol),于100 ℃~110 ℃(回流)反应4 h。倒入5%盐酸(15 mL)中,用乙醚(3×30 mL)萃取以除去非碱性物质;水层加入过量50%氢氧化钠溶液后用乙醚(3×30 mL)萃取;合并醚层,用无水硫酸镁干燥;蒸干溶剂得灰褐色油状液体2a1.47 g,收率86%, b.p.(120~126) ℃/80 Pa。
将1b0.32 g(1.35 mmol)溶于三氯氧磷(5 mL)中,搅拌下于110 ℃(回流)反应4 h。减压除尽三氯氧磷,残余物用饱和NaHCO3溶液调至pH为中性,用CH2Cl2(3×20 mL)萃取;合并有机液,用饱和食盐水洗涤,无水Na2SO4干燥;蒸干溶剂,残余物经硅胶柱层析[洗脱液:A=V(石油醚) ∶V(乙酸乙酯)=50 ∶1]分离纯化得白色固体2b0.30 g,收率88.2%, m.p.162 ℃~163 ℃;1H NMRδ: 4.04(s, 2H), 7.48~7.50(m, 2H), 7.52~7.61(m, 2H), 7.72~7.76(t,J=7.24 Hz, 8.02 Hz, 1H), 8.18~8.26(m, 3H);13C NMRδ: 28.98, 117.17, 118.44, 120.04, 120.27, 121.35, 122.35, 124.16, 124.42, 125.25, 127.81, 132.59, 134.86, 139.08, 143.82, 156.10; IRν: 3 032, 2 883, 1 625, 1 557, 1 502, 1 398, 1 370, 1 252, 1 117, 920, 836, 759, 722, 618 cm-1; HR-MS: Calcd for C16H10NCl 251.050 2, found 251.050 2。
(3) 3的合成
在反应瓶中依次加入2a1.47 g(8.6 mmol),硝基苯5 mL,二甲苯10 mL和10%Pd/C 120 mg,搅拌下回流反应9 h(油水分离器收集水)。冷却至室温,过滤;滤液用5%盐酸(3×30 mL)萃取,乙醚洗涤;水层用固体碳酸钾调至pH≈9后用乙醚(3×30 mL)萃取;合并醚层,用无水硫酸镁干燥;蒸干溶剂,残余物经硅胶柱层析(洗脱液:A=50 ∶1)分离纯化得黄色固体5H-茚[1,2-b]吡啶(3a)1.26 g,收率87.7%, m.p.93 ℃~95 ℃;1H NMRδ: 8.60(d,J=4.84 Hz, 1H), 8.12(d,J=7.38 Hz, 1H), 7.82(d,J=7.57 Hz, 1H), 7.48(ddd,J=27.84 Hz, 19.01 Hz, 7.27 Hz, 1H), 7.31~7.12(m, 1H), 3.88(s, 1H);13C NMRδ: 34.68, 121.00, 121.25, 125.33, 127.47, 128.84, 132.54, 136.85, 140.99, 143.78, 148.35, 160.56; IRν: 3 037, 2 921, 1 567, 1 450, 1 413, 1 395, 1 171, 773, 752, 622 cm-1; HR-MS: Calcd for C12H9N 167.073 5, found 167.073 5。
将2b0.30 g(1.20 mmol)溶于THF(30 mL)中,搅拌下加入10%Pd/C 60 mg,在氢气压(50 psi)下于室温反应15 h。过滤,滤液用饱和NaHCO3调至pH中性;用CH2Cl2(2×20 mL)萃取,合并有机层,用饱和食盐水洗涤,无水Na2SO4干燥;蒸干溶剂后用无水乙醇重结晶得白色针状晶体11H-茚[1,2-b]喹啉(3b)0.23 g,收率89.0%, m.p.162 ℃~163 ℃;1H NMRδ: 4.06(s, 2H), 7.49~7.54(m, 3H), 7.61~7.63(m, 1H), 7.69~7.73(t,J=7.66 Hz, 1H), 7.83~7.85(d,J=8.11 Hz, 1H), 8.21~8.23(d,J=8.18 Hz, 2H), 8.32~8.34(d,J=7.08 Hz, 1H);13C NMR δ: 156.56, 142.92, 140.05, 135.23, 129.53, 126.08, 124.91, 123.97, 123.76, 122.73, 122.44, 122.30, 120.59, 120.40, 116.98, 28.92; IRν: 3 060, 2 924, 1 625, 1 562, 1 500, 1 393, 1 320, 1 119, 961, 895, 771, 735, 618 cm-1; HR-MS: Calcd for C16H11N 217.089 1, found 217.089 2。
1.3 5的合成
将3a0.15 g(0.9 mmol)溶于干燥的CH2Cl2(5 mL)中,N2保护下加入三氟甲磺酸甲酯0.21 mL(1.8 mmol),搅拌下于室温反应22 h(TLC跟踪)。蒸干溶剂,残余物经硅胶柱层析[洗脱液:B=V(CH2Cl2) ∶V(甲醇)=100 ∶1~50 ∶1作梯度洗脱]分离纯化得白色固体1-甲基-5H-茚[1,2-b]吡啶三氟甲磺酸盐4a0.28 g,收率94%。
将4a0.28 g(0.85 mmol)与对二甲氨基苯甲醛0.19 g(1.27 mmol)在冰醋酸(50 mL)中回流反应2 d。减压蒸馏,残余物经柱层析(洗脱液:B=100 ∶1)分离纯化得红色粉末5a0.26 g,收率67%, m.p.201 ℃~203 ℃;1H NMR(DMSO-d6) δ: 3.05(s, 6H, NMe2,Z-构型), 3.09(s, 6H, NMe2,E-构型), 4.71(s, 3H, CH3), 6.88(d,J=8.31 Hz, 2H),7.62~7.72(m, 4H), 7.78~8.01(m, 1H), 8.33~8.46(m, 3H), 8.84~8.89(m, 1H), 9.09(d,J=7.72 Hz, 1H);13C NMR(DMSO-d6) δ: 36.3, 52.4(×2), 84.8, 117.2(×2), 126.5(×2), 128.7(×2), 128.9(×2), 131.3(×2), 133.5, 134.1, 136.0, 137.4(×2), 138.3(×2), 140.0(×2), 141.7, 143.5, 144.2, 144.7(×2), 147.6, 148.7(×2), 152.7, 155.4, 157.3(×2); IRν: 3 096, 2 906, 1 615, 1 580, 1 526, 1 455, 1 435, 1 362, 1 263, 1 222, 1 153, 1 031, 943, 903, 823, 729, 638 cm-1。
用类似的方法合成深红色固体5b,收率65.2%, m.p.176 ℃~178 ℃;1H NMR(DMSO-d6)δ: 3.07(s, 6H, NMe2,Z-构型), 3.10(s, 6H, NMe2,E-构型), 4.86(s, 3H, CH3), 6.82~6.89(m, 2H), 7.61~7.97(m, 5H), 8.15~8.37(m, 4H), 8.62~8.70(m, 2H), 9.33~9.57(d,J=7.8 Hz, 1H);13C NMR(DMSO-d6) δ: 34.6, 46.4(×2), 117.1(×2), 124.3(×2), 126.6(×2), 128.3, 128.7(×2), 132.2, 132.6, 133.6(×2), 134.0(×2), 134.6, 136.0(×2), 136.8, 137.8, 138.4, 139.4(×2), 139.5, 140.0(×2), 141.5(×2), 142.0, 143.5(×2), 146.9, 151.0, 157.1, 157.6, 159.9(×2); IRν: 3 090, 2 922, 2 852, 1 620, 1 590, 1 525, 1 370, 1 343, 1 263, 1 223, 1 190, 1 156, 1 029, 945, 814, 758, 636 cm-1。
1.4 5的体外抗肿瘤活性测定
以HEP-2细胞(人喉鳞癌细胞)和K562细胞(人红白血病细胞)为测试细胞株,采用MTT法对5进行了抗肿瘤活性实验。取对数生长期的肿瘤细胞,离心后用RPMI 1640培养液稀释成5×104个·mL-1,接种于96孔板中,37 ℃, 5 %CO2培养24 h后加入不同浓度的样品,再孵化48 h;加入10μL MTT溶液( 5μg·mL-1),孵化4 h后,每孔加入100μL酸化的10%SDS,在酶标仪上570 nm处测定吸收度值(630 nm为参照波长),按公式{抑制率=(对照组OD值-治疗组OD值)/对照组OD值×100%}计算被测物对癌细胞生长的抑制率,以改良寇氏法计算IC50值。
2 结果与讨论
对于茚并吡啶类和茚并喹啉类化合物的三氟甲磺酸盐衍生物的合成路线,首先形成骨架5H-茚[1,2-b]吡啶和11H-茚并[1,2-b]喹啉,再形成它们的三氟甲磺酸盐衍生物。文献报道5H-茚[1,2-b]吡啶中间体的合成路线有多种[4~8],本文最终选定的5H-茚[1,2-b]吡啶的合成路线为:以1-茚酮为原料,制备烯胺、环合、脱氢芳构化合成5H-茚[1,2-b]吡啶。将其与三氟甲磺酸甲酯成盐,再与对二甲氨基苯甲醛偶联得5a。
一些文献[9~11]报道了11H-茚并[1,2-b]喹啉中间体的合成。本课题组以1-茚酮为原料,经熔融法、卤代、还原合成11H-茚并[1,2-b]喹啉,甲基化、偶联得5b。
2.1 合成
因为烯胺不稳定,易水解,因此在反应时必须用氮气保护且严格除水,减压抽出后立刻投入下一步反应;2a,4a和4b的合成需在严格的无水条件下操作;通过熔融法制备1b反应时间较文献缩短到1.5 h;在3b的合成中通过使用重结晶纯化的方法得产物3b,收率较高,避免了繁琐的柱层析操作;氮上的甲基化反应,我们以二氯甲烷代替文献[9,11]的甲苯作为溶剂,不但降低了毒性,而且4a和4b的产率更高,都达95%左右;5a和5b的合成采用在醋酸中回流,与文献[12,13]方法的醇和醇钠或者乙酸酐条件相比,条件简单,且产率更高;由核磁共振氢谱图可以看出5a和5b均为两种顺反异构体的混合物。5a中吡啶环电子云密度小,Z-构型NMe2化学位移向高场移动为3.05;E-构型则相反,向低场移动为3.09,且根据峰面积分析E-构型占绝大多数,同时出现双倍碳数;5b中喹啉环电子云密度小,分析同前,Z-构型和E-构型峰面积相当,并出现双倍碳数。
2.2 抗肿瘤活性
5a和5b的抗肿瘤活性测试结果见表1。由表1可见,5a和5b均表现出很好的抗肿瘤活性。对HEP-2的IC50值分别为1.37μg·mL-1和3.96μg·mL-1,对K562的IC50值分别为0.94μg·mL-1和1.30μg·mL-1,抑制活性强。

表 1 5a和5b的抗肿瘤活性*
*HEP-2:人喉鳞癌细胞;K562:人红白血病细胞; 测试方法见1.4
3 结论
在茚并吡啶骨架和在其基础上引入苯环取代基的茚并喹啉环都有助于化合物的抗肿瘤活性。该类化合物具有广阔的应用前景,关于此类化合物的构效关系及进一步的生物活性测试结果将陆续报道。
[1] Katayama H, Kawada Y, Oshiyama T,etal. Synthetic inhibitors of DNA topoisomerase Ⅰ and Ⅱ[J].Chem Pharm Bull,1999,47(1):48-53.
[2] Katayama H, Kiryu Y, Kaneko K,etal. Anticancer activities of pyrazolo[1,5-a]indole derivatives[J].Chem Pharm Bull,2000,48(11):1628-1633.
[3] Zhu Y M, Qin L N, Liu R,etal. Synthesis of pyrazolo[1,5-a]indoles via copper(Ⅰ)-catalyzed intramolecular amination[J].Tetrahedron Lett,2006,48:6262-6266.
[4] Parcell R F, Hauck F P. The preparation of tetrahydropyridines from enamines and imines[J].J Org Chem,1963,28:3468-3469.
[5] Curt W, Angelika D, Werner R,etal. Intramolecular cyclization of nitrile imines.Synthesis of indazoles,fluorenes,and aza analogues[J].J Org Chem,1978,43(10):2037-2041.
[6] Akio O, Takayuki K, Hiroshi I,etal. Flash vacuum pyrolysis of substituted pyridineN-oxides and its application to syntheses of heterocyclic compounds[J].J Org Chem,1982,47(18):3497-3503.
[7] Mark T DuPriest, Charles L, Daniel K,etal. A facile synthesis of 7-halo-5H-indeno[1,2-b]pyridines and pydin-5-ones[J].J Org Chem,1986,51(11):2021-2023.
[8] Milton Wolf, Chester P. Thietano[3,2-a]indan-1,1-dioxides[P].US 3 632 579A1,1972.
[9] Xue Y Zhu, Leroy G Mardenborough, Shouming Li,etal. Synthesis and evaluation of isosteres ofN-methyl indolo[3,2-b]-quinoline (cryptolepine) as new antiinfective agents[J].Bioorganic & Medicinal Chemistry,2007,15:686-695.
[10] Hans V M, Nele L, Bart A,etal. Improved ruthenium catalysts for the modifed friedlaender quinoline synthesis[J].New J Chem,2007,31:1572-1574.
[11] Donald E Bierer, Larisa G Dubenko, Pingsheng Zh-ang,etal. Antihyperglycemic activities of cryptolepine analogues:An ethnobotanical lead structure Isolated from cryptolepis sanguinolenta[J].J Med Chem,1998,41:2754-2764.
[12] Paver G V, Pavel K G, Tilichenko M N. Synthesis and some properties of substituted 4-azafluorenes[J].Chemistry of Heterocyclic Compounds,1990,26(7):950-953.
[13] Prostakov N S, Soldatenkov A T, Radzhan P K,etal. Synthesis and transfomations of 1-methyl-4-azafluorene[J].Chemistry of Heterocyclic Compounds,1982,18(4):390-394.
