橙色红曲菌乳清苷-5'-磷酸脱羧酶基因的克隆
2010-11-10王伯华李燕萍
王伯华,许 杨,,*,李燕萍
(1.南昌大学食品科学与技术国家重点实验室,江西南昌330047;2.南昌大学中德联合研究院,江西南昌330047)
橙色红曲菌乳清苷-5'-磷酸脱羧酶基因的克隆
王伯华1,许 杨1,2,*,李燕萍2
(1.南昌大学食品科学与技术国家重点实验室,江西南昌330047;2.南昌大学中德联合研究院,江西南昌330047)
根据已报道的真菌乳清苷-5′-磷酸脱羧酶(pyrG)氨基酸序列,采用CODEHOP策略设计简并引物,从橙色红曲菌(Monascus aurantiacus)的基因组DNA中克隆得到pyrG基因片段。然后运用PCR法筛选橙色红曲菌Fosmid文库,获得了pyrG基因全长(GenBank登录号:GU723506)。经过blastx比较发现,橙色红曲菌pyrG基因与Aspergillus flavus NRRL3357的氨基酸序列同源性最高,达到81%。该基因能够作为筛选标记应用于橙色红曲菌同源转化系统。
橙色红曲菌,简并引物,pyrG基因,基因克隆
乳清苷-5′-磷酸脱羧酶是尿嘧啶核苷酸合成途径中的一种关键性酶,缺乏该酶的营养缺陷株不能在不含尿嘧啶的培养基上生长。通过基因转化,向其导入含pyrG基因的质粒,可使其恢复在基础培养基上生长的能力。如果将要导入的基因连接在该质粒上,pyrG可作为外源基因是否导入的标志[1]。红曲菌(Monascus)是我国传统的酿造微生物,能产生多种有益的代谢产物:红曲色素[2]、Monacolin类化合物[3]、γ-氨基丁酸[4]等。近年来,欧美等国对红曲菌的研究兴趣也在不断增加[5-6]。本实验采用CODEHOP( Consensus - Degenerate Hybrid Oligonucleotide Primer)PCR结合 PCR筛选红曲菌Fosmid文库的方法克隆了橙色红曲菌pyrG基因全长,该基因的获得为橙色红曲菌同源转化系统的建立奠定了基础,也为红曲菌及其它真菌的新基因克隆提供了一个有效的方法。
1 材料与方法
1.1 材料与仪器
橙色红曲菌AS3.4384 购自中国科学院微生物研究所;E.coli DH5α 购自Novagen公司;橙色红曲菌Fosmid文库 由本实验室李燕萍博士构建;Taq酶、T4连接酶、pMD18-T载体、限制性内切酶、质粒提取试剂盒及胶回收试剂盒 均购自TaKaRa公司;引物 Invitrogen公司合成。
1.2 实验方法
1.2.1 简并引物的设计 根据在NCBI上已经公布的真菌pyrG基因的氨基酸序列,采用CODEHOP[7]策略设计一对简并引物:

1.2.2 pyrG基因片段的扩增 采用氯化苄法从橙色红曲菌菌丝体中提取基因组DNA[8],并以此为模板,用OMP-F和OMP-R引物进行简并PCR扩增,PCR产物进行 TA克隆,初步筛选阳性克隆子后送Invitrogen公司测序,得到橙色红曲菌pyrG基因片段序列。

表1 橙色红曲菌pyrG基因与其它真菌的同源性比较
1.2.3 PCR法筛选橙色红曲菌Fosmid文库 根据测序所得pyrG基因片段序列设计一对特异性引物,用于筛选Fosmid文库:

橙色红曲菌Fosmid文库保存于21块384微孔板中,将每一块孔板的384个克隆子混合培养并提取质粒DNA,分别命名为Q1~Q21,作为PCR法筛选文库的模板DNA。当确定阳性克隆子所在的孔板后,分别提取阳性孔板中的每行(编号为A~P)和每列(编号为1~24)的质粒DNA,用作PCR方法定位单克隆子的模板DNA。进一步用PCR验证单克隆子后提取其质粒DNA备用。
1.2.4 OMPD基因全长的获得 用各种限制性内切酶对阳性Fosmid单克隆子质粒进行酶切,将1~5kb的酶切片段分别进行回收、克隆、测序。采用DNASTAR软件对测序结果进行拼接,获得pyrG基因全长序列。
2 结果与分析
2.1 pyrG基因片段的克隆与分析
以橙色红曲菌AS3.4384基因组DNA为模板,OMP-F和OMP-R为简并引物进行PCR反应,在PCR反应退火温度选择上,从47~63℃设置了温度梯度,PCR产物经1%琼脂糖凝胶电泳检测,在431bp的位置出现目的条带(图1)。由图1可以看出,退火温度低于60℃时会产生许多非特异性条带,当退火温度达到63℃时出现特异性目的条带。
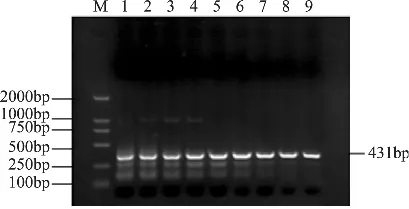
图1 不同退火温度下简并PCR扩增结果
简并PCR的特异性产物TA克隆后送公司测序。测序结果应用blastx程序进行比对,比对结果表明,由克隆所得基因片段推测的氨基酸序列具有pyrG基因的典型保守区域,可以基本确认所获得的DNA片段是橙色红曲菌pyrG基因的部分序列。
2.2 橙色红曲菌Fosmid文库的筛选
以Q1~Q21板池混合质粒作为模板,OMP-S1和OMP-S2为引物进行PCR筛选,能够扩增pyrG目的条带的有:Q6、Q12、Q17。然后制备Q12板的行池(A~P)和列池(1~24),以这些小混合池作为模板,采用PCR反应分别扩增pyrG基因片段,F行、9列能够扩增出目的条带。根据筛选结果,可初步确定阳性克隆子为Q12F9,提取Q12F9单克隆子质粒作为模板,采用 PCR反应对其进一步验证,能够观察到360bp的目的条带(图2)。

图2 PCR法筛选橙色红曲菌Fosmid文库
2.3 pyrG基因全长的克隆与分析
选用几种常用的限制性内切酶对Q12F9质粒进行酶切,酶切结果如图3。将1~5kb的酶切片段分别进行回收、克隆,经PCR验证含有pyrG基因片段的阳性克隆子送公司测序。测序结果拼接后获得了pyrG基因全长。通过blastx检测与其它真菌的同源性大小(表 1),发现橙色红曲菌 pyrG基因与Aspergillus flavus NRRL3357的pyrG氨基酸同源性达到81%,说明克隆所得基因为pyrG基因。
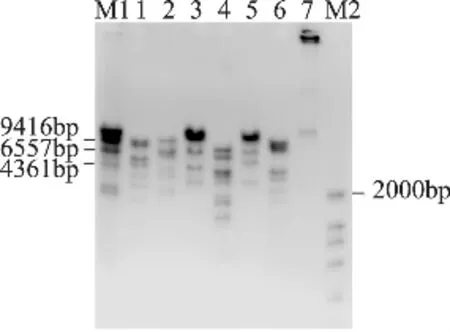
图3 限制性内切酶酶切Q12F9电泳图
3 讨论
传统简并引物设计思路是对与目标基因亲缘关系比较近的氨基酸序列进行对比后,根据其保守序列和密码子的简并性设计出长度在18~23bp的引物序列。由于传统引物很少考虑到物种的密码子偏好性,简并度较高,同时传统简并引物的Tm值一般都比较低,这些因素往往导致PCR结果不理想,特异性不高,假阳性率增加。本研究通过CODEHOP策略来设计简并引物,引物3′端为核心简并区,5′端为非简并性夹板结构,5′端夹板结构是根据不同物种密码子偏好性设计的特异性序列[9]。它大大减少了引物的简并度且不影响引物的有效性,与传统方法设计的简并引物相比,具有扩增特异性高、PCR反应条件易摸索的优点。
常规筛选基因组文库的方法是通过菌落杂交或将排列在微孔板中的单克隆复制到尼龙膜上筛选阳性克隆[10-11],费时费力。Green[12]首先提出了采用PCR方法筛选混合池快速定位阳性克隆子,为筛选单克隆减少了很大的工作量,避免了人力和物力的浪费。
本研究采用CODEHOP PCR结合PCR筛选橙色红曲菌Fosmid文库的方法克隆了橙色红曲菌pyrG基因全长,为红曲菌及其它真菌新基因的克隆提供了一个有效的途径。该基因全长的获得为橙色红曲菌同源转化系统的构建奠定了基础。
[1]Hartingsveldt W,Mrttern I E,Zeijl C M J,et al.Development of a homologous transformation system for Aspergillus niger based on the pyrG gene[J].Mol Gen Genet,1987,206:71-76.
[2]Johanna F G,Jurgen D,Lothar L.Use of Monascus extracts as an alternative to nitrite in meat products[J].Fleischwirtsch,1991,71(10):1184-1186.
[3]Heber D,Yip I,Ashley J M,et al.Cholesterol lowering effects of a proprietary Chinese red-yeast-rice dietary supplement[J].A M J Chin Nuer,1999,69:231-236.
[4]Kono I,Himeno K.Antimicrobial activity of Monascus pilosus IFO 4520 against contaminant of Koji[J].Biosci Biotechnol Biochem,1999,63:1494-1496.
[5]Blanc P J,Laussac J P,Bars J L,et al.Characterization of monascidin A from Monascus as citrinin[J].Int J Food Microbiol,1995,27:201-213.
[6]Campoy S,Pérez F,Martín J F,et al.Stable transformation of the azaphilone pigment-producing Monascus purpureus obtained by protoplast transformation and Agrobacterium-mediated DNA transfer[J].Curr Genet,2003,43:447-452.
[7]Rose T M,Henikoff J G,Henikoff S.CODEHOP(Consensus-Degenerate Hybrid Oligonucleotide Primer)PCR primer design[J].Nucleic Acids Research,2003,31:3763-3766.
[8]袁勇芳,许杨,李思光,等.红曲霉DNA提取及其RAPDPCR反应体系的建立[J].生物技术,2005,18:9-14.
[9]Rose T M,Schultz E R,Henikoff J G,et al.Consensusdegenerate hybrid oligonucleotide primers for amplification of distantly related sequences[J].Nucleic Acids Research,1998,26(7):1628-1635.
[10]Ioannou P A,Amemiya C T,Garnes J,et al.A new bacteriophage P1-derived vector for the propagation of large human DNA fragments[J].Nature Genet,1994(6):84-89.
[11]Zhang H B,Choi S,Woo S S,et al.Construction and characterization oftwo rice bacterialartificialchromosome libraries from the parents of a permanent recombinant inbred mapping population[J].Mol Breed,1996(2):1-14.
[12]Green E D,Olson M V.Systematic screening of yeast artificial chromosome libraries by use of the polymerase chain reaction[J].Proc Natl Acad Sci USA,1990,87:1213-1217.
Cloning of Orotidine-5′-phosphate decarboxylase gene from Monascus aurantiacus
WANG Bo-hua1,XU Yang1,2,*,LI Yan-ping2
(1.State key laboratory of Food Science and Technology,Nanchang University,Nanchang 330047,China;2.Sino-Germany Joint Research Institute,Nanchang University,Nanchang 330047,China)
According to the known Orotidine-5′-phosphate decarboxylase,pyrG gene fragment was cloned from Monascus aurantiacus genomic DNA using degenerate primers designed with CODEHOP strategy.And its complete sequence was obtained by a PCR-based method for screening fosmid library(Accession No.GU723506). After the blastx comparison,the cloned gene was confirmed that it was Monascus pyrG gene with 81%sequence identity to that of Aspergillus flavus NRRL3357.lt can use as a selective marker for Monascus aurantiacus homologus transformation system.
Monascus aurantiacus;degenerate primer;pyrG gene;gene cloning
Q785
A
1002-0306(2010)11-0172-03
2010-04-20 *通讯联系人
王伯华(1981-),女,博士研究生,主要从事食品生物技术方向的研究。
国家自然科学基金(30860123)。
