芳烃类污染物结构与水溶性(-lgSw)关系研究
2010-09-25廖立敏李建凤雷光东
廖立敏,朱 俊,李建凤,雷光东*
(1.内江师范学院资源与环境科学学院,四川内江641112;2.内江师范学院化学与生命科学学院,四川内江替换为 641112)
芳烃类污染物结构与水溶性(-lgSw)关系研究
廖立敏1,2,朱 俊1,李建凤1,2,雷光东1*
(1.内江师范学院资源与环境科学学院,四川内江641112;2.内江师范学院化学与生命科学学院,四川内江替换为 641112)
为探寻芳烃类有机物结构与水溶性(-lgSw)的关系,应用分子顶点及顶点相互作用值对芳烃类有机物进行了结构表征.采用多元线性回归及逐步回归建立了2个定量结构-性质关系的-lgSw模型.经过比较,发现模型(M2)具有最佳模拟结果,此时模型的复相关系数(R)为0.949,标准偏差(SD)为0.443.采用Jackknife法对模型进行了稳健性检验,结果表明回归模型具有可接受的总体稳健性及良好的预测能力;另外,采用留一法(leave-one-out)对模型进行交叉检验,复相关系数(Rcv)为0.915,标准偏差(SDcv)为0.566,这也说明所建模型的稳定性与预测能力均较为理想.
环境化学;芳烃;水溶性;分子顶点;定量结构-性质相关
随着化学工业的迅猛发展及人们生活水平的日益提高,大量人工合成芳烃类化合物进入环境中,其中一些为有害化合物,严重地威胁着生态环境和人类健康.芳烃类有机污染物是重要的环境污染物,因此对其性质进行研究具有一定的意义.溶解度(Sw)是化合物的重要物理参数,不仅对化学反应的顺利进行具有重要意义,而且在环境化学中也是研究有机污染物环境归趋时的重要参数.虽然通过实验测定芳烃类化合物的溶解度(Sw)仍是目前最为有效的方法,但该法费时费力.通过对化合物结构与性质之间关系的深入认识,对大量的有机物性质进行预测显示出广泛的应用前景[1-5].本文选取34个芳烃类化合物(实验数据取自文献[6],列于表1)为研究对象,寻找化合物结构与其水溶性(-lgSw)之间的关系模型.
对化合物的结构进行表征时,忽略非骨架氢原子的影响,将非氢原子(骨架原子)看成是分子的顶点.根据顶点原子的连接性特征将顶点原子进行分类,再考虑到不同类型的顶点原子自身杂化状态及顶点原子之间的电性相互作用对分子性质的影响构建分子结构表征方法(本文暂时将该表征方法称为“顶点及顶点相互作用值”).采用多元线性回归及逐步回归的方法,对芳烃的水溶性(-lgSw)进行了定量相关性研究,发现芳烃水溶性(-lgSw)与分子结构参数之间存在良好的线性关系.

表1 34个芳烃类化合物及其水溶性(-lgSw)Tab.1 34 compounds and their-lgSw

续 表
1 原理与方法
在有机化合物的分子隐氢图中,每一个非氢原子即为分子顶点.通常认为分子顶点本身所处状态及顶点之间的相互作用对整个有机物分子的性质有重要的影响,而分子中的非骨架原子(氢原子)对整个分子性质的影响可以忽略不计.并且处于不同连接状态下的顶点及顶点之间的相互作用对分子性质的贡献可能不同,同类型的顶点及顶点之间的相互作用对分子性质的贡献相似且具有加和性.因此首先将分子内的顶点按照其连接状态进行分类,分子内的顶点依据其所连接的其它顶点数分为A1、A2、A3、A4 四类 ,分别表示与 1、2、3、4 个其它顶点相连,如与三个顶点相连的叔碳原子属于A3顶点类型.
对于分子顶点本身所处状态对分子性质的贡献,本文引入了 Hall和 Kier[7]所提出的原子固有状态(I),并对文献的计算方法进行了一定的修正,得到了所谓的分子顶点值,用于表征分子顶点自身的状态对分子性质的影响.其计算式如下:

上式中,xr表示分子中顶点类型为r的所有顶点i的贡献值,νi是顶点i的原子价电层的电子数;ni为该顶点价电层所属的主量子数;δiσ+π是该顶点参与成σ和π键的总电子数;δiσ为成σ键的电子数.如单个CSP、C2SP、C3SP在分子中的顶点贡献值分别是2.5000、1.6667、1.2500.上述定义比 Hall和 Kier原始定义多个系数 (ν/4)1/2且δσ+π和δσ值计算中没有减去该原子与氢成键电子数,即重点放在顶点原子杂化状态表达上.根据顶点原子的分类,分子中最多含四类顶点原子,则每个分子将最终得到四个顶点值,分别记为 x1、x2、x3和 x4.
对于分子顶点间的相互作用对分子性质的贡献,顶点之间的相互作用与顶点原子电性及相隔距离密切相关.一般而言,依据库仑定律,电性越大、距离越小,相互作用就越大.本文借鉴文献[8-10]的表征方法来表征分子顶点间的相互作用对分子性质的贡献,即化合物分子中四类顶点发生相互作用可以组合出以下几种方式:m11、m12、m13、m14、m22、m23、m24、m33、m34、m44,分别记为 x5、x6、x7、x8、x9、x10、x11、x12、x13、x14十个顶点相互作用值.其计算式如下:
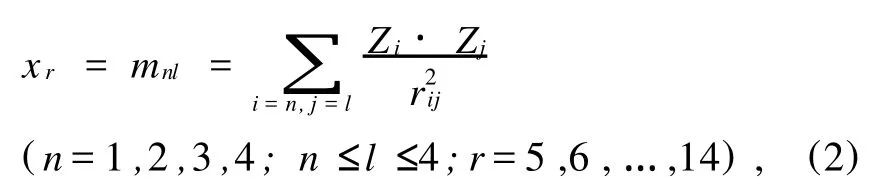
上式中,n和l为顶点所属类型;Zi、Zi为顶点i、j相对于C原子的相对电负性,如氯原子的相对电负性为3.16/2.55=1.2392;rij是顶点i、j之间的相对距离(即所经最短途径键长之和与碳碳单键键长的比值).
依据以上原理,对于每一个有机化合物分子,最多将有十四个变量来描述其结构信息.所有样本分子的十四个结构参数值,见表1(由于所有样本分子中不含第 4 类原子 ,所以 x4、x8、x11、x13、x14为全零项,在表1中已经删除).
2 建模与分析
多元线性回归(multiple linear regression,MLR)是一种经典的建模方法,它对自变量和因变量加以线性拟合以得到最小二乘(least square,LS)意义下的最佳结果.采用SPSS13.0中的多元线性回归(MLR)方法对表1中各变量和-lgSw(Exp)进行相关性研究,同时采用留一法对模型进行交叉检验,得到9变量的预测模型(M1)的相关结果如下:

N为样本数,R为复相关系数,SD为标准偏差,F为显著性检验值;Rcv为交叉检验复相关系数,SDcv为交叉检验的标准偏差,Fcv为显著性检验值(下同).
此模型复相关系数达到0.951,说明模型拟合效果较好.但是模型变量数为9,样本数只有34,即模型不符合“样本数/变量数≧5”的经验规则,说明变量数过多,有可能存在过拟合现象.采用SPSS13.0统计学软件包对M1进行回归诊断,分别计算出9个变量的 t统计值及方差膨胀因子(VIF).通过分析发现模型M1的确具有一定程度多重共线性(某些变量VIF值偏大),而且并不是所有变量都表现出显著特征(部分变量 t值落在-2≤t≤2范围).为了进一步考察各变量对建模过程的影响,消除模型可能存在的过拟合现象以进一步提高模型的质量,对变量进行了逐步回归(SMR)分析,同时采用留一法对模型进行交叉检验,模型复相关系数(R)、交叉检验的复相关系数(Rcv)、标准偏差(SD)及交叉检验的标准偏差(SDcv)随变量引入的变化情况见图1.
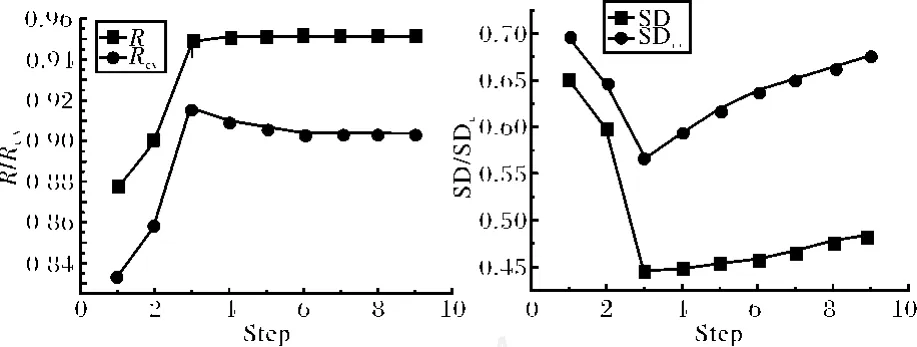
图1 逐步回归R及SD的变化曲线Fig.1 Plot ofRand SD change with the step of SMR
从图1可以看到,当3个变量入选模型时复相关系数(R)接近最大值,交叉检验的复相关系数(Rcv)达到最大值,同时标准偏差(SD)及交叉检验的标准偏差(SDcv)达到最小值,综合考虑 R、Rcv、SD、SDcv,应该选择3个变量进行建模.3变量的预测模型(M2)如下:
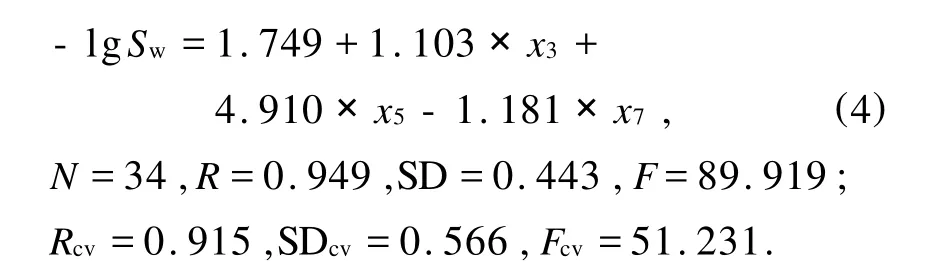
上述模型建模效果较好,且模型符合“样本数/变量数≧5”的经验规则.M2与M1相比,复相关系数(R)仅降低0.002,而变量数由9个减少到3个,大大降低了模型的复杂程度.再次对M2进行回归诊断发现3个变量的t绝对值均大于2(其中最小为5.128),而VIF值表现为显著降低(最大为30.962),从而证实该模型具有较高的质量.
为说明模型(M2)的稳定性和可靠性,采用Jackknife[9]法对模型进行了稳健性检验.每次从34个化合物中去掉化合物序号中个位数分别是0,1,2,…,9的分子,剩余化合物作为建模组,根据方程 (4)的形式进行回归.重复10次,所得Jackknifed的 R和SD值列于表2.
其中 R值最大为0.965、最小为0.931,平均值为0.949.SD值最大为0.465、最小为0.356,平均值为0.440.从这十组数据可以看出,相关系数及标准偏差均波动不大,基本上呈正态分布,说明回归模型具有可接受的总体稳健性及良好的预测能力.另外,“留一法”对模型(M2)进行交叉检验的复相关系数(Rcv)值为0.915,这比原模型的复相关系数(R)值0.949略低;交叉检验标准偏差(SDcv)值为0.566,也只是比原模型的标准偏差(SD)值0.443略大,这也说明所建模型的稳定性与预测能力均较为理想.
方程(4)中的 x3表示化合物第三类顶点原子自身状态值,x5表示第一类原子与第一类原子相互作用值,x7表示第一类原子与第三类原子相互作用值,因此第一类及第三类原子对该样本集化合物的水溶性(-lgSw)有较大的影响.即苯环上的取代基越多、分布越紧密、末端原子电性越强,化合物的水溶性(-lgSw)值就越大.17号化合物苯环上取代基最多、并且分布紧密、末端原子为Cl原子(电负性较强),因而该化合物的水溶性(-lgSw)值最大.另外,14、15、32号化合物,取代基也较多,也有类似表现.模型(M2)对34个化合物的水溶性(-lgSw)的估计值列于表1中-lgSw(Cal.)栏.模型对某些样本的预测误差过大,可能是由于其本身的分子结构特殊性所造成的,也可能是实验数据本身就存在某些误差,从而影响计算结果.当然也有可能是分子结构表征方法不够完善,本方法是基于分子的二维平面结构,而分子的实际结构是三维的.这些问题在今后的研究中都应该予以考虑.

表2 模型(M2)的稳健性检验Tab.2 Robustness test of the model(M2)
3 结论
本文将分子顶点及顶点间的相互作用值对分子性质的贡献用于34个芳烃类化合物水溶性(-lgSw)的研究,取得的结果满意.本文分子结构表征法与现今流行的三维分子建模法如比较分子场法(CoMFA)[3,4]相比,不需要考虑构象的叠加,计算相对较为简便.运用多元线性回归结合逐步回归构建了2个预测模型,经比较发现模型(M2)具有最佳的模拟结果.模型经Jackknife法和交叉验证法检验,具有可接受的总体稳健性及良好的预测能力,用该模型对34个芳烃类化合物的水溶性(-lgSw)进行了模拟,模拟结果与实验结果间的误差较小.模型(M2)对芳烃类化合物水溶性(-lgSw)具有一定的预测能力,在缺乏实验数据的情况下,模型的预测值具有一定的参考价值.
[1]隆兴兴,牛军峰,史姝琼.邻苯二甲酸酯类化合物正辛醇-水分配系数的QSPR研究 [J].环境科学,2006,27(11):2318-2322.
[2]廖立敏,梅 虎,郑怀礼,等.大气中痕量挥发性有机物的结构表征和保留时间的估计与预测 [J].环境化学,2007,26(6):838-840.
[3]杨 嵩,万 坚,陈 婷,等.α-氧代膦酸酯类衍生物的CoMFA研究[J].华中师范大学学报:自然科学版,2006,40(1):58-59,74.
[4]万 坚,张青叶,刘瑞华,等.HMG-CoA还原酶抑制剂的3D-QSAR研究[J].华中师范大学学报:自然科学版,2005,39(4):487-5489.
[5]杨光富,杨华铮.嘧啶(硫)醚类除草剂的非线性QSAR研究[J].华中师范大学学报:自然科学版,2001,35(1):30-34.
[6]王连生,韩朔睽.有机污染化学进展[M].北京:化学工业出版社,1995:14-17.
[7]Hall L H,Kier L B.Electrotopological state index for atom types:a novel combination of electronic,topological,and valence state information[J].J Chem Inf Comput Sci,1995,35:1039-1045.
[8]Limin Liao,Hu Mei,Jianfeng Li,et al.Estimation and prediction on retention times of components from essential oil of paulownia tomentosa flowers by molecular electronegativity distance vector[J].J Mol Struct(THEOCHEM),2008,850:1-8.
[9]Lili Sun,Liping Zhou,Yu Yu,et al.QSPR study of polychlorinated diphenyl ethers by molecular electronegativity distance vector(MEDV-4)[J].Chemosphere,2007,66:1039-1051.
[10]Shushen Liu,Chunsheng Yin,Shaoxi Cai,et al.Molecular structural vector description and retention index of polycyclic aromatic hydrocarbons[J].Chemom Intell Lab Syst,2002,61:3-15.
Abstract:In order to explore the relationship between water-solubility and structure of aromatic compounds,values of molecular vertexes and their interaction were proposed to describe the structures of aromatic compounds.Through multiple linear regression(MLR)and stepwise regression,two models of-lgSwwere obtained.By comparison,model(M2)showed better results of simulation.The correlation coefficient(R)of the model was 0.949 and the standard deviation(SD)of the model was 0.443.Jackknife method was used to test its stability,the results showed that the regression model with acceptable stability and good predictive ability.In addition,the model was evaluated by performing the cross-validation with the leave-one-out(LOO)procedure.The cross-verification multiple correlation coefficient(RCV)and the standard deviation(SDCV)were 0.915 and 0.566,respectively.This also showed that the stability and predictive ability of the model was desirable.
Key words:environmental chemistry;arene;water-solubility;structural descriptors;quantitative structure-property relationship
Study on quantitative structure-water-solubility relationship of aromatic compounds
LIAO Limin1,2,ZHU Jun1,LI Jianfeng1,2,LEI Guangdong1
(1.College of Resource and Environment Science,Neijiang Normal University,Neijiang,Sichuan 641112;2.College of Chemistry and Life Science,Neijiang Normal University,Neijiang,Sichuan 641112)
O641;O625
A
1000-1190(2010)04-0610-04
2010-05-29.
四川省教育厅青年基金资助项目(09ZB036);四川省科技厅应用基础项目(2008J Y0121).
*通讯联系人.E-mail:leiguangd@163.com;liaolimin523@126.com.
