香果健消片的质控方法研究Ⅱ:木香主要成分的鉴别和含量测定
2010-09-22申力文李夸巧孙艳芬肖培云
申力文,李夸巧,孙艳芬,肖培云
(大理学院药学院,云南大理 671000)
香果健消片的质控方法研究Ⅱ:木香主要成分的鉴别和含量测定
申力文,李夸巧,孙艳芬,肖培云*
(大理学院药学院,云南大理 671000)
目的:建立香果健消片中木香主要成分的鉴别及含量测定方法。方法:采用薄层色谱法鉴别木香;高效液相色谱法测定木香烃内酯和去氢木香内酯的含量。结果:薄层色谱法鉴别斑点明显,木香烃内酯和去氢木香内酯进样量在0.10~2.00μg范围内与峰面积的线性关系良好。加样回收率分别为99.3%(RSD=1.24%,n=6)和100.8%(RSD=1.67%,n=6)。结论:上述方法专属性强、重复性好,可用于木香的鉴别、木香中木香烃内酯及去氢木香内酯的含量测定。
香果健消片;薄层色谱法;高效液相色谱法;木香烃内酯;去氢木香内酯
香果健消片收载于《卫生部药品标准(WS3-B-1585-93)》,由蜘蛛香、草果、木香等药材制备而成,具有健胃消食之功效〔1〕。其中木香具有行气止痛、健脾消食的作用。现代研究表明木香的主要活性成分为木香烃内酯和去氢木香内酯〔2〕,应为含木香制剂质量控制的主要指标。为保证该制剂的内在质量,对处方中木香进行了薄层色谱法(TLC)鉴别,并采用高效液相色谱法(HPLC)对木香烃内酯和去氢木香内酯进行了定量研究。
1 仪器与试药
1.1 仪器 高效液相色谱系统(美国Agilent 1200);AL204-IC电子天平(梅特勒-特利多仪器有限公司);薄层色谱成像仪(武汉药科新技术开发有限公司);超声波清洗机(宁波新芝生物科技股份有限公司)。
1.2 试药 香果健消片(云南云河药业有限公司,批号:081201、090603、090706、090802);木香烃内酯、去氢木香内酯对照品(批号分别为111524-200804、111525-200706,由中国药品生物制品检定所提供),木香对照药材(批号:120921-200506);薄层层析硅胶G(青岛海洋化工有限公司);甲醇为色谱纯;水为超纯水;其他试剂为分析纯。
2 方法和结果
采用TLC进行鉴别,HPLC进行定量。具体方法如下。
2.1 木香鉴别
2.1.1 供试液的制备 取香果健消片20片,除去糖衣,研细,精密称取2.5 g,加乙醚30 mL,搅匀,静置10min,超声处理30min,滤过,滤液置水浴上低温挥干,残渣加乙酸乙酯1mL,使其溶解,作为供试品〔3〕。
2.1.2 对照药材溶液的配制 精密称定0.5 g木香药材,加乙醚6mL,摇匀,静置10min,超声处理30min,过滤,滤液置水浴上低温挥干,残渣加乙酸乙酯0.5mL使其溶解,即得对照药材溶液。
2.1.3 对照品溶液的配制 分别取木香烃内酯对照品与去氢木香内酯对照品,精密称取10.0mg,于10mL容量瓶中,加甲醇溶解、定容、摇匀,作为储备液。精密移取上述溶液1mL于10mL容量瓶中,加甲醇定容、摇匀,即得0.10mg/mL的混合对照品溶液。
2.1.4 阴性对照品溶液的制备 按处方制备缺木香的阴性对照品,再按“2.1.1”项制备,即得阴性对照品溶液。
2.1.5 薄层鉴别结果 用毛细管吸取供试品、对照药材、木香烃内酯对照品、去氢木香内酯对照品、阴性对照品溶液,分别点于同一硅胶G薄层板上,以甲苯-乙酸乙酯-冰醋酸(15∶1∶0.1)〔4〕为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液〔3-7〕,在105℃加热至斑点显色清晰,日光下检视。供试品色谱中,在与对照药材色谱及对照品色谱相应的位置上,显相同颜色的斑点,阴性对照无干扰。见图1。
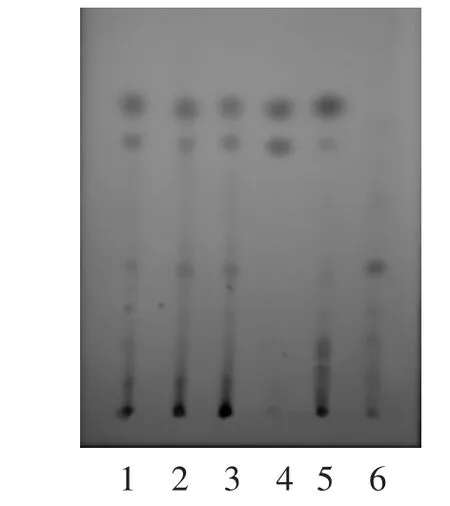
图1 木香的TLC图谱
2.2 木香烃内酯和去氢木香内酯的含量测定
2.2.1 色谱条件 色谱柱为Agela Venusil XBP-C18柱(250 mm×4.6 mm,5 μm);流动相为甲醇-水(70∶30)〔3,5,8-10〕;检测波长为220 nm;柱温为25 ℃〔3,5,9-10〕;流速为1.0 mL/min,进样5 μL。
2.2.2 供试液的制备 取本品20片,去糖衣,精密称定,研细,取约0.5 g粉末,精密称定,精密加入甲醇20mL,称定,25℃下超声提取40min,放冷,补重,摇匀,用微孔滤膜滤过,取续滤液,即得。
2.2.3 阴性对照品溶液的制备 取缺木香的阴性对照品0.5 g,按“2.2.2”项供试品溶液的制备方法制备,即得。
2.2.4 专属性试验 取对照品溶液、供试品溶液和阴性对照品溶液各5μL进样测定,结果供试品色谱中呈现与对照品保留时间相同的色谱峰,阴性对照品在与木香烃内酯、去氢木香内酯对照品相同的保留时间处未显色谱峰,故认为无干扰。见图2-4。
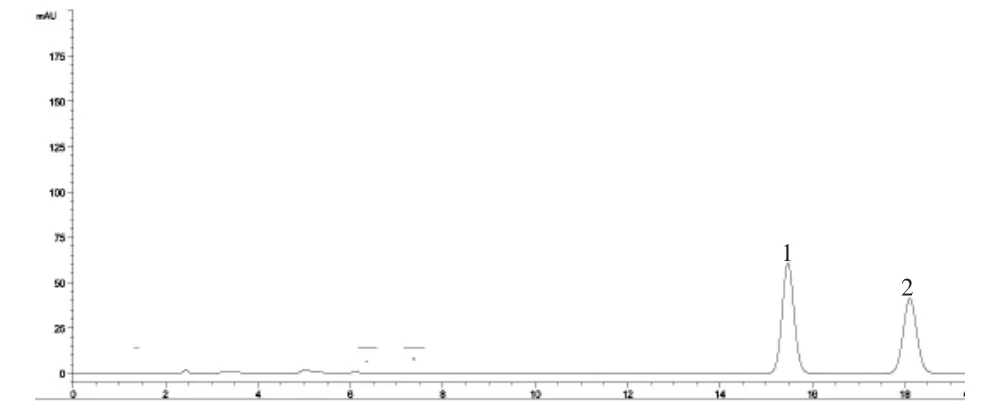
图2 对照品HPLC色谱图
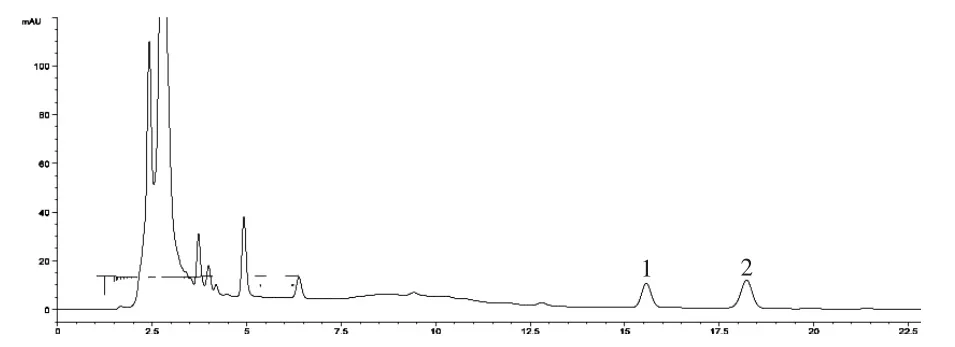
图3 供试品溶液HPLC色谱图
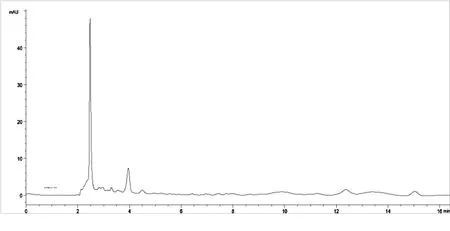
图4 阴性对照品HPLC色谱图
2.2.5 线性关系考察 精密移取0.10 mg/mL对照品混合液1,2,5,10,12,15,20 μL,分别注入高效液相色谱仪,测定峰面积值。结果对照品峰面积Y与其进样量X(μg)之间呈良好的线性关系,木香烃内酯和去氢木香内酯的线性范围均为0.1~2.0μg,回归方程分别为
2.2.6 精密度试验 精密移取对照品混合液,连续进样6次,测其峰面积,木香烃内酯和去氢木香内酯的峰面积的RSD分别为0.31%和0.24%,结果表明精密度良好。
2.2.7 稳定性试验 精密移取同一供试品溶液,按上述色谱条件在0,3,5,18,20,24 h进样测定,得木香烃内酯和去氢木香内酯的RSD分别为2.01%和0.78%。结果表明样品溶液在24 h内稳定。
2.2.8 重复性试验 取同一批号(批号:090603)样品,按供试品溶液制备项下操作,平行制备6份,按上述色谱条件测定,木香烃内酯和去氢木香内酯平均含量分别为0.118 2mg/g和0.214 2mg/g,RSD分别为1.57%和0.85%。结果表明重复性良好。
2.2.9 加样回收率试验 精密称取已知含量的香果健消片6份,按表1精密加入对照品溶液,按供试品溶液制备项操作,按上述色谱条件测定,计算回收率,结果见表1。
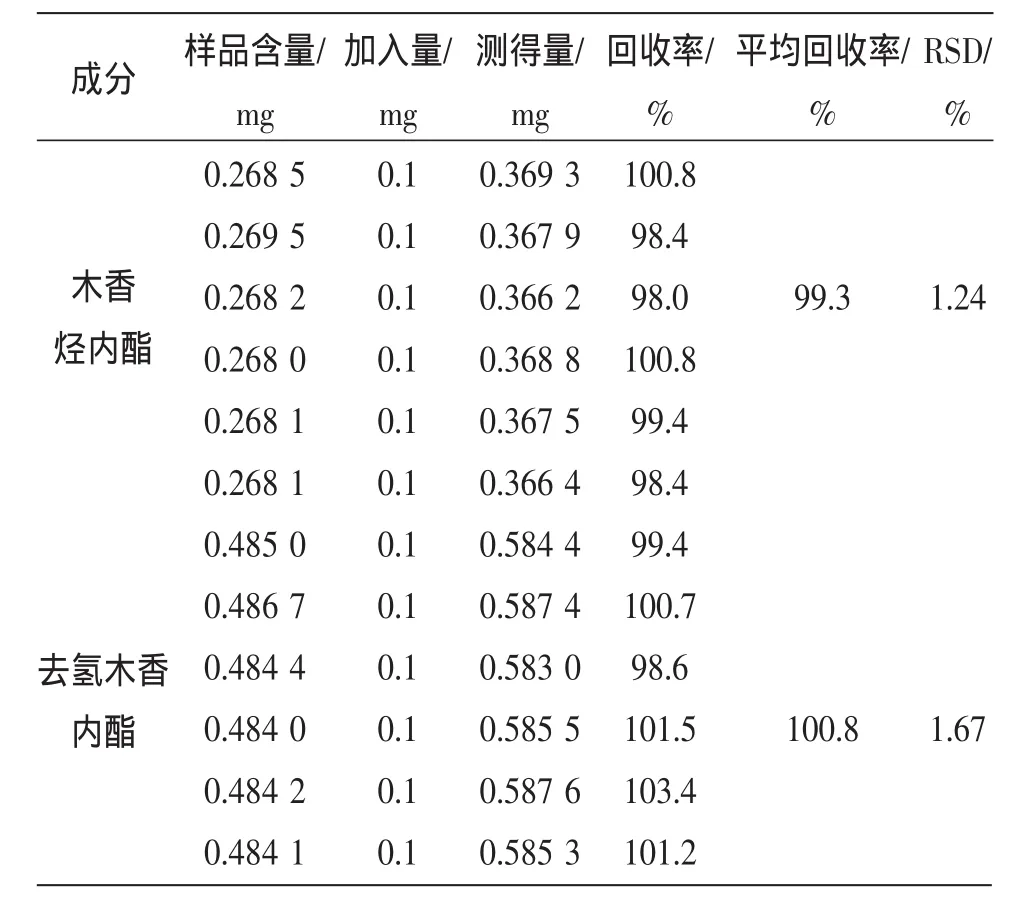
表1 回收率实验结果(n=6)
2.2.10 样品含量测定 取4个不同批号的样品,按供试品溶液制备方法操作,按上述色谱条件测定并计算结果。见表2。
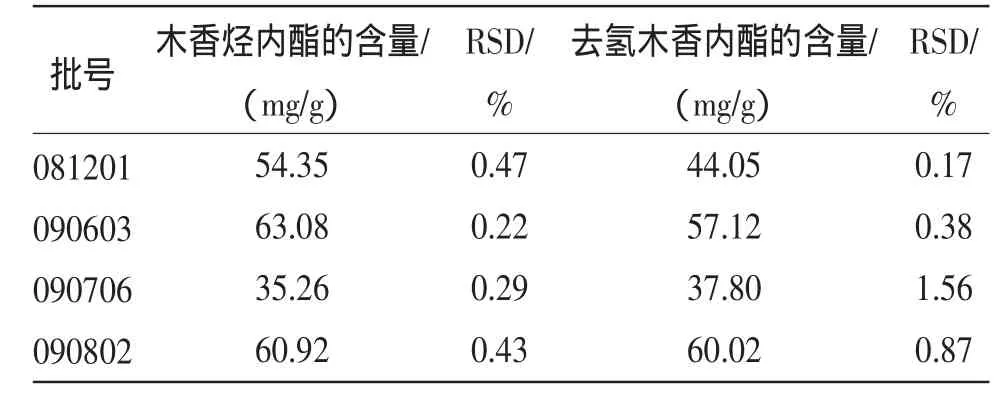
表2 木香烃内酯和去氢木香内酯的含量测定(n=3)
3 讨论
3.1 提取时间考察 本实验对提取时间进行了考察,分别考察了20,30,40,50,60 min,结果表明40min即可提取完全,故选择提取时间为40min。
3.2 TLC条件的选择 在TLC实验中考察了环己烷-丙酮(10∶3)〔3〕、环己烷-丙酮-甲苯(10∶3∶1、10∶3∶2)、甲苯-乙酸乙酯-冰乙酸(10∶1∶0.1、12∶1∶0.1、14∶1∶0.1、15∶1∶0.1等)的展开效果,其中甲苯-乙酸乙酯-冰乙酸(15∶1∶0.1)分离效果最好,故选用该展开系统。
3.3 HPLC条件的选择 试验中考察了甲醇-水(65∶35、70∶30、72∶28、75∶25等)的分离效果,结果表明甲醇-水(70∶30)的分离效果最佳;同时还考察了不同柱温,以25℃最为适宜。
本试验采用TLC薄层色谱法对香果健消片中木香进行了薄层鉴别,实验结果表明阴性对照液对供试液无干扰,专属性强,色谱清晰,特征斑点分离良好,斑点无拖尾现象,可以作为该药品定性鉴别的指标。本试验建立了同时测定香果健消片中木香烃内酯和去氢木香内酯的方法,方法可靠,专属性强。
〔1〕国家药典委员会.中华人民共和国卫生部药品标准〔M〕.北京:人民卫生出版社,1990:120.
〔2〕王永兵,王强,毛福林,等.木香的药效学研究〔J〕.中国药科大学学报,2001,32(2):146.
〔3〕蔡丽云,李子鸿,李怀国,等.胃舒颗粒质量研究标准〔J〕.中国药业,2007,16(13):20-21.
〔4〕高咏莉.木香理气片的质量标准研究〔J〕.天津药学,2007,19(4):18-19.
〔5〕严行开,何泽民.HPLC法测定五味香连丸中木香烃内酯和去氢木香内酯的含量〔J〕.今日药学,2008,18(6):35-37.
〔6〕王荣红,俞嘉,熊志立,等.得生丸质量标准研究〔J〕.中国药房,2010,21(15):1387-1388.
〔7〕姜娟,焦海胜,周素琴,等.七味散质量标准的研究〔J〕.中国药师,2008,11(3):297-298.
〔8〕余晓琴,李萍,董林,等.九香止痛丸的鉴别及木香烃内酯和去氢木香烃内酯的测定〔J〕.华西药学杂志,2009,24(3):312-313.
〔9〕戚巍,周凯,王晔.枫香脂十味丸中木香烃内酯和去氢木香内酯的含量测定〔J〕.中国民族医药杂志,2006(6):67-68.
〔10〕王晔,高沂.八味三香散质量标准中鉴别和含量测定方法的研究〔J〕.中国药学,2007,42(3):232-233.
(责任编辑 蒋 康)
The Quality ControlMethod of Xiangguojianxiao TabletsⅡ:Identifying and Determ ining the Main Com ponents of Costustroot in Xiangguojianxiao Tablets
SHEN Liwen,LIKuaqiao,SUN Yanfen,XIAO Peiyun*
(College of Pharmacy,Dali University ,Dali,Yunnan 671000,China)
Objective:To establish a method for identification of Costustroot and determination of costunolide and dehydrocostus lactone in Xiangguojianxiao tablets.Methods:Costustroot was identified by TLC;the contents of costunolide and dehydrocostus lactone were determined by HPLC.Results:The developed TLC spots were quite clear;the calibration curve of costunolide and dehydrocostus lactone were linear within 0.10-2.00μg.The average recoveries of costunolide and dehydrocostus lactone were 99.3%with RSD of 1.24%(n=6)and 100.8%with RSD of 1.67%(n=6),respectively.Conclusion:The established methods are specific,reproducible and suitable for the identification of costustrootand determination of costunolide and dehycostus lactone.
Xiaoguojianxiao Tablets;TLC;HPLC;costunolide;dehydrocostus lactone
R944.4
B
1672-2345(2010)12-0007-03
大理学院大学生科研基金资助项目(2009DXSY01)
2010-06-13
申力文,主要从事药物分析研究.
*通信作者:肖培云,副教授,电话:15808729786.
