蟾乌巴布膏的质量标准研究
2010-09-17周庆氢
周庆氢, 张 聪, 谢 松
(1.上海雷允上药业有限公司,200002;2.上海市中药研究所,201401)
蟾乌巴布膏是刘嘉湘教授的临床验方,由蟾蜍、川乌、重楼等24味名贵药材组成,应用现代制剂技术,采用亲水性高分子辅料制备的巴布膏。蟾乌巴布膏对肿瘤以及其他疼痛均有良好的疗效,更有活血化瘀,消肿止痛之功效,被临床广泛认可,是国内唯一肿瘤中药外用药。为控制该制剂质量,采用色谱法对其中的蟾酥、生川乌、生关白附、两面针、荜茇、细辛、乳香、没药、丁香、肉桂等药材进行了鉴别,并采用气相谱法对蟾乌巴布膏中樟脑、薄荷脑、冰片、水杨酸甲酯等多个有效成分的含量进行了测定。
1 仪器与试药
1.1 仪器 HP-6890 plus气相色谱仪(美国HP公司),氢火焰检测器,HS3120型超声波提取仪(淮阴汉邦科技有限公司),梅特勒托利多AE240电子天平(十万分之一)。
1.2 试药 丁香酚、桂皮醛、胡椒碱、樟脑、薄荷脑、冰片、水杨酸甲酯对照品,两面针、荜茇、细辛、乳香、没药对照药材均购自中国药品生物制品检定所,石油醚(60~90℃)为分析纯(杭州炼油厂),其余试剂均为分析纯(中国医药集团上海化学试剂公司),硅胶G、硅胶H、硅胶GF254薄层预制板(青岛海洋化工厂)。
1.3 样品 蟾乌巴布膏(上海雷允上药业有限公司),批号:071001,071101,071201。
2 方法与结果
2.1 色谱鉴别
2.1.1 蟾酥的薄层鉴别[1]取蟾乌巴布膏1片,剪成小块,除去盖衬,置150 mL回流瓶中,加氯仿50 mL,加热回流1 h,取上清液蒸干,残渣加氯仿1 mL使溶解,作为供试品溶液。
另取脂蟾毒配基及华蟾酥毒基对照品,加氯仿制成每1 mL含1 mg的溶液,作为对照品溶液。
照薄层色谱法(中国药典2005年版一部附录VI B)试验[2],吸取供试品溶液及对照品溶液各5 μL分别点于同一硅胶G薄层板上,以环己烷-氯仿-丙酮(4∶3∶3)为展开剂,展开,取出,晾干,喷以10%磷钼酸乙酸溶液,105℃加热至斑点显色清晰,供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点(见图1)。
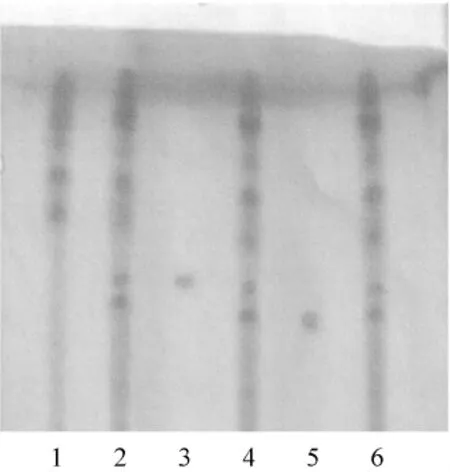
图1 蟾酥薄层鉴别色谱图
2.1.2 生川乌、生关白附的薄层鉴别[3]取蟾乌巴布膏1片,剪成小块,除去盖衬,置150 mL回流瓶中,加浓氨试液0.5 mL,氯仿50 mL,加热回流1 h,取上清液蒸干,残渣加无水乙醇1 mL使溶解,作为供试品溶液。
取生川乌、生关白附对照药材1 g,加浓氨试液0.5 mL,氯仿 50 mL,加热回流 1 h,放冷,滤过,滤液蒸干,残渣加无水乙醇1 mL使溶解,作为对照药材溶液。
另取乌头碱对照品,加无水乙醇制成每1 mL含1 mg的溶液,作为对照品溶液。
照薄层色谱法(中国药典2005年版一部附录VI B)试验,吸取供试品溶液15μL,及对照药材、对照品溶液各10μL分别点于同一硅胶G薄层板上,以环己烷-二乙胺(8∶2)为展开剂,展开,取出,晾干,喷以稀碘化铋钾,供试品色谱中,在与对照药材、对照品色谱相应的位置上,显相同颜色的斑点。
2.1.3 两面针的薄层鉴别[4]取蟾乌巴布膏1片,剪成小块,除去盖衬,置150 mL回流瓶中,加浓氨试液0.5 mL,氯仿50 mL,加热回流1 h,取上清液蒸干,残渣加氯仿1 mL使溶解,作为供试品溶液。
另取两面针对照药材1 g,加浓氨试液0.5 mL,氯仿30 mL,加热回流1 h,放冷,滤过,滤液蒸干,残渣加氯仿1 mL使溶解,作为对照药材溶液。
照薄层色谱法(中国药典2005年版一部附录VI B)试验,吸取供试品溶液5μL,对照药材溶液3 μL,分别点于同一硅胶G薄层板上,以苯-乙酸乙酯-甲醇-异丙醇-浓氨试液(20∶5∶3∶1∶0.12)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。
2.1.4 荜茇的薄层鉴别[5]取蟾乌巴布膏1片,剪成小块,除去盖衬,置150 mL回流瓶中,加浓氨试液0.5 mL,氯仿50 mL,加热回流1 h,取上清液蒸干,残渣加氯仿1 mL使溶解,作为供试品溶液。
取荜茇对照药材0.5 g,加浓氨试液0.5 mL,氯仿30 mL,加热回流1 h,放冷,滤过,滤液蒸干,残渣加氯仿1 mL使溶解,作为对照药材溶液。再取胡椒碱对照品,加无水乙醇制成每1 mL含1 mg的溶液,作为对照品溶液。
照薄层色谱法(中国药典2005年版一部 附录VI B)试验,吸取供试品溶液10μL,对照药材1μL和对照品溶液5μL,分别点于同一硅胶G薄层板上,以苯-乙酸乙酯-丙酮(7∶2∶1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇(v/v)溶液,105℃加热至斑点显色清晰,置紫外光灯(254 nm)下检视。供试品色谱中,在与对照药材、对照品色谱相应位置上,显相同颜色的荧光斑点(见图2)。
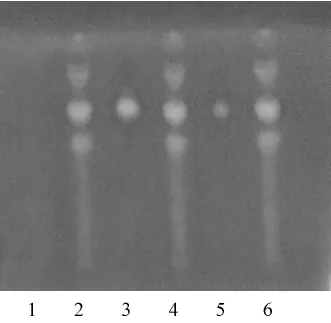
图2 荜茇的薄层鉴别色谱图
2.1.5 细辛的薄层鉴别[6]取本品1片,剪成小块,除去盖衬,置150 mL回流瓶中,加乙酸乙酯50 mL,加热回流1 h,取上清液蒸干,残渣加乙酸乙酯1 mL使溶解,作为供试品溶液。
另取细辛对照药材1 g,加乙酸乙酯30 mL,加热回流1 h,滤过,滤液蒸干,残渣加乙酸乙酯1 mL使溶解,作为对照药材溶液。
照薄层色谱法(中国药典2005年版一部附录VI B)试验,吸取供试品溶液10μL和对照药材溶液3μL,分别点于同一硅胶G薄层板上,以石油醚(60~90℃)-乙酸乙酯(85∶15)为展开剂,展开,取出,晾干,喷以10%磷钼酸乙醇溶液,105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。(见图3)

图3 细辛的薄层鉴别色谱图
2.1.6 乳香、没药的薄层鉴别[7]取本品1片,剪成小块,除去盖衬,置150 mL回流瓶中,加乙酸乙酯50 mL,加热回流1 h,取上清液蒸干,残渣加乙酸乙酯1 mL使溶解,作为供试品溶液。
取乳香、没药对照药材各0.5 g,各加乙酸乙酯30 mL,加热回流1 h,滤过,滤液蒸干,残渣分别加乙酸乙酯2 mL使溶解,作为对照药材溶液。
照薄层色谱法(中国药典2005年版一部附录VI B)试验,取供试液溶液10μL,对照药材溶液各1 μL,分别点于同一硅胶G薄层板上,以环己烷-乙酸乙酯(85∶15)为展开剂,展开,取出,晾干,喷以10%磷钼酸乙醇溶液,105℃加热至斑点显色清晰,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。(见图4)

图4 乳香、没药的薄层鉴别色谱图
2.1.7 丁香、肉桂的鉴别 取本品2片,剪成小块,除去盖衬,置250 mL回流瓶中,加乙酸乙酯100 mL,水浴上回流1 h,迅速冷却,用铺有无水硫酸钠的漏斗滤过,滤液减压回收至干,残渣用乙酸乙酯溶解并转移至2 mL量瓶中,用乙酸乙酯稀释至刻度,离心,取上清液,作为供试品溶液。
另取丁香酚和桂皮醛对照品,分别加乙酸乙酯制成每1 mL含丁香酚0.15 mg、桂皮醛0.015 mg的溶液,作为对照品溶液。照气相色谱法(中国药典2005年版一部附录VI E)试验,用弹性石英毛细管柱,以聚乙二醇PEG-20M为固定相,柱温为180℃。分别吸取供试品溶液和对照品溶液各1μL,注入气相色谱仪。供试品溶液色谱图中应呈现与对照品保留时间相同的色谱峰。
2.2 樟脑、薄荷脑、冰片、水杨酸甲酯含量测定[8]
2.2.1 试液的配制 内标溶液的制备:取萘适量,精密称定,加无水乙醇溶解,制成每1 mL含1.2 mg的溶液,即得。
混合标准品的制备:取樟脑、薄荷脑、冰片和水杨酸甲酯对照品适量,分别精密称定,分别加无水乙醇制成每1 mL含1、2、1、1 mg的溶液。分别精密量取上述4种对照品溶液1 mL,置同一20 mL量瓶中,精密加入上述内标溶液1 mL,加无水乙醇稀释至刻度,摇匀,即得。
供试品溶液的制备:取蟾乌巴布膏5片,每片分别精密截取6 cm2,共30 cm2,剪成小块,除去盖衬,置150 mL回流瓶中,精密加入无水乙醇50 mL,称定重量,置水浴上回流2 h,迅速用冰浴冷却,用无水乙醇补足减失的重量,滤过,精密量取续滤液10 mL,置20 mL量瓶中,精密加入内标溶液1 mL,加无水乙醇稀释至刻度,摇匀,即得。
2.2.2 色谱条件及系统适应性试验 J&W INNOWAY弹性石英毛细管柱(固定相为聚乙二醇PEG-20 m,30 m ×0.53 mm,固定相厚度 1 μm),程序升温,柱温140℃保持23 min,以每分钟6℃升温至200℃保持20 min,进样温度280℃;检测器温度300℃,分流进样,分流比1∶1,理论板数以薄荷脑计算不低于20 000。
分别取樟脑、薄荷脑、冰片、水杨酸甲酯空白样品进行平行试验,空白样品均无干扰,且供试品在内标物保留时间处无干扰。见图5~7。

图5 供试品中樟脑、薄荷脑、冰片、水杨酸甲酯含量测定气相色谱图

图6 基质空白溶液气相色谱图
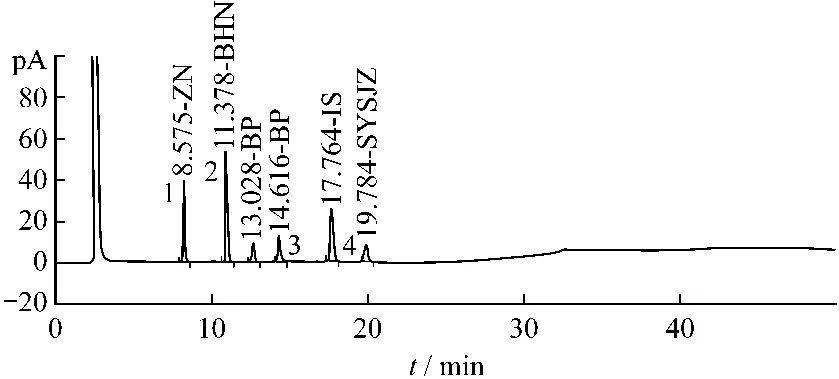
图7 樟脑、薄荷脑、冰片、水杨酸甲酯对照品溶液气相色谱图
2.2.3 线性关系试验 精密吸取混合对照品贮备液按一定稀释比例,配制成不同浓度的混合对照品溶液,分别吸取1μL注入气相色谱仪,测得各组份及内标物的峰面积,并计算回归方程,结果见表1。

表1 樟脑、薄荷脑、冰片、水杨酸甲酯等线性关系考察
表明:在线性范围内樟脑、薄荷脑、冰片、水杨酸甲酯线性关系良好。
2.2.4 日间精密度 取线性关系试验中取同一供试品溶液在不同日分别进样5次,每次1μL,在以下时间点:0、1、2、3、5 d 时测定色谱峰吸收值,结果樟脑、薄荷脑、冰片、水杨酸甲酯的 RSD分别为0.88% 、1.65% 、1.42% 、1.11% 。
2.2.5 日内精密度 取线性关系试验中同一供试品溶液在当日分别进样5次,每次1μL,在以下时间点:0、1、2、4、10 h 时测定色谱峰吸收值,结果樟脑、薄荷脑、冰片、水杨酸甲酯的 RSD分别为0.32% 、1.00% 、0.29% 、1.49% 。
2.2.6 重复性试验 取蟾乌巴布膏(批号:071001),分取5份,分别按2.2.1项及2.2.2项下的方法制备供试品溶液,进样测定,结果见表2。

表2 日重复性试验(n=5)
2.2.7 回收率试验 取蟾乌巴布膏(批号:071101),分取6份,加入一定量对照品,按2.2.1项及2.2.2项下的方法制备供试品溶液,进样测定,结果见表3。

表3 回收率实验结果(n=6)
2.2.8 样品测定 分别精密吸取3批样品(071001、071101、071201)的供试品溶液,与混合对照品溶液1μL注入气相色谱仪,进样测定含量,结果见表4。
3 讨论
3.1 蟾乌巴布膏是采用亲水性高分子辅料制备的巴布膏,为保证产品质量我们对该产品中的多味药材进行了定性的研究,采用薄层色谱法对其中的蟾酥、生川乌、生关白附、两面针、荜茇、细辛、乳香、没药、丁香、肉桂等药材进行了鉴别,建立蟾乌巴布膏中蟾酥、两面针、荜茇、细辛、乳香、没药、丁香、肉桂的定性方法,其中生川乌和生关白附的鉴别,因为存在干扰,故需要进一步研究。

表4 样品的含量测定
3.2 本试验比较了以不同溶剂(甲醇、无水乙醇)、不同提取方法(超声、回流、索氏提取)和不同超声时间(15、30、45、60 min)对樟脑、薄荷脑、冰片和水杨酸甲酯提取效果的影响。结果以无水乙醇回流提取的效率最高,回流时间2 h,已可将樟脑、薄荷脑、冰片和水杨酸甲酯提取完全,故选用无水乙醇回流提取2 h作为提取条件。
3.3 通过实验,制定了蟾乌巴布膏中蟾酥、两面针、荜茇、细辛、乳香、没药、丁香、肉桂等八味药材的色谱鉴别方法,并用气相色谱法测定了产品中樟脑、薄荷脑、冰片、水杨酸甲酯的含量。方法简便、专属性强、重现性好,可有效的控制蟾乌巴布膏的质量。
[1]白 雪,张 莉,齐 刚.蟾酥的鉴别检查和含量测定方法[J].时珍国医国药,2004,(4):226-227.
[2]中国药典[S].一部,2005.
[3]夏昌隆,张 璇,薛 洁.伤科黑药膏中乌头碱的薄层色谱鉴别[J].新疆中医药,2001(S1).
[4]招荣鑑,孙亦群,彭 彤.祛风通络散中大黄与两面针的薄层色谱鉴别及酯型生物碱的限量检查[J].时珍国医国药,2006,(7):1258-1259.
[5]原永芳,卞 俊.超临界流体萃取法在筚茇质量控制中的应用[J].中国药科大学学报,2000,(3):199-202.
[6]赵瑞芝,袁小红,丘小惠.疣毒净霜剂的薄层鉴别方法[J].中国药业,2006,15(2):61-62.
[7]蒋 敏,段海燕.接骨七厘片中血竭、乳香、没药的薄层色谱鉴别[J].湖南中医学院学报,2000,(4):30-31.
[8]陈振霆.气相色谱法测定无极膏中樟脑、薄荷脑、水杨酸甲酯、麝香草酚的含量[J].海峡药学,2005,(3):65-67.
