非质量硫同位素分馏效应研究进展
2010-09-14马红梅李院生安春雷
马红梅,李院生,姜 苏,安春雷
(中国极地研究中心极地科学国家海洋局重点实验室,上海 200136)
非质量硫同位素分馏效应研究进展
马红梅,李院生,姜 苏,安春雷
(中国极地研究中心极地科学国家海洋局重点实验室,上海 200136)
非质量硫同位素分馏效应是目前国际上最前沿的稳定同位素地球化学研究领域之一。在简要介绍非质量分馏理论的基础上,对近几年非质量硫同位素分馏效应的最新研究成果进行总结和分析。关于非质量硫同位素分馏的微观来源机制存在较多争议,有待于进一步探索;采用SF6为工作气体是现有硫同位素高精度测定的主要制样技术;非质量硫同位素分馏效应研究为火星大气演化及火星生命痕迹探询、古代大气氧化条件、地球早期硫循环、火山活动对气候的影响等重大地质科学问题的解释开辟了一条独特的新途径。最后对非质量硫同位素分馏领域研究趋势进行了探讨。
硫同位素;分馏效应;来源机制;制样技术;地质学;研究趋势
0 引言
硫广泛存在于宇宙和自然界中。在宇宙中,硫是第10位最丰富的元素;在地壳中,硫是第14位最丰富的元素[1]。硫总共有4种稳定同位素(32S、33S、34S、36S),其丰度分别为95.02%、0.75%、4.21%、0.02%[2]。硫同位素比值可以用于判别硫源和追索其运移途径,对于了解地球表面氧化还原环境和探索地球历史演化具有重要意义。
长期以来,学者多认为自然界中各种过程所引起的硫同位素分馏均属于与质量相关的分馏过程,即绝大部分硫化物和硫酸盐的硫元素的稳定同位素比值相对标准(标样)相应比值的千分偏差δ(33S)、δ(34S)和δ(36S)之间存在着定量关系: δ(33S)=0.515×δ(34S);δ(36S)=1.9×δ(34S)[3]。2000年以来,地球科学家们相继在火星陨石、月球尤其是地球物质中发现“非质量硫同位素分馏效应”(mass independent sulfur isotope fractionation)存在的证据,并将这一现象与太阳系早期大气成分及其演化、古代大气氧化条件、地球各圈层的相互作用、地球早期硫循环等一系列重大地球科学问题联系起来,为许多重要假说的解释提供了一个新思路。随着硫检测技术精度不断提高以及同位素分馏理论的不断完善,围绕着自然界非质量硫同位素分馏的物理--化学起源、高精度硫同位素组成分析方法的建立及其应用展开了较为深入的研究,非质量硫同位素分馏研究也成为国际上最前沿的同位素地球化学研究领域之一。
1 定义与表示方法
同位素分馏是自然界中最常见的现象,通常由物理、化学和生物作用引起的热力学和动力学同位素分馏,几乎均为与质量相关的分馏。1965年, Hulston等[3]通过分析不同类型陨石中各种硫化物4种稳定的硫同位素,发现δ(33S)、δ(34S)、δ(36S)之间存在一个固定的比例关系(图1),这与物理-化学过程中的同位素分馏理论一致。Hulston[4]通过计算机模拟得出更加精确的推算方程

式中:δ(XS)中X为33、34和36。
凡是不遵循上述质量分馏定律的现象称为非质量同位素分馏。非质量硫同位素分馏效应的大小通常用Δ(33S)和Δ(36S)表示,即
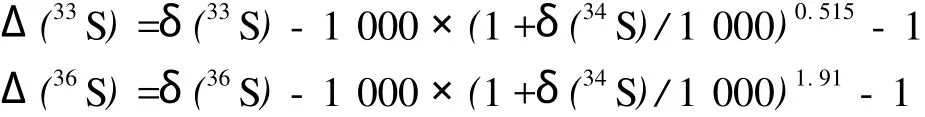
当Δ(33S)≠0和Δ(36S)≠0时,认为具有硫同位素非质量分馏效应。目前,用作计算Δ(33S)和Δ(36S)的硫同位素标准物质是凯尼昂迪布洛陨硫铁(Canyon Diablo Troilite,CDT),它被认为代表地球原始的硫同位素组成[5],其绝对值N(34S)/N(32S)为0.045,δ(34S)为0[6]。由于近30 a来的使用,CDT已经缺乏,国际原子能机构推荐Vienna Canyon Diablo Troilite(VCDT)替代作为硫同位素标准物质[7]。
2 研究进展
2.1 来源与保存
2.1.1 非质量硫同位素来源机制
目前已经在陨石[8-11]、太古代沉积物[12-15]、南非钻石[16]、气溶胶[17]、南极冰芯[18-19]中发现了非质量硫同位素分馏现象。关于非质量硫同位素分馏效应如何产生并能在地球样品中得到保存,前人展开了较为深入的探讨。具有代表性的观点有:

图1 陨石和地质样品中δ(33S)、δ(36S)和δ(34S)之间的关系Fig.1 Relationships Amongδ(33S),δ(36S)andδ(34S) in Meteorite and Terrestrial Samples
(1)20世纪60年代,Hulston等[8]从不含陨硫铁微粒(硫质量分数小于0.01%)的陨石铁相中分解出硫,并检测到了富33S和36S(δ(36S)高达(6.3~21)×10-3)的现象,推测是宇宙射线与铁相互作用时所产生的碎裂反应所致。
(2)Clayton等[20]对陨石硫同位素组成分析认为,陨石中非质量硫同位素分馏效应是核事件过程中保存了早期太阳系同位素组成不均一特征引起的。
(3)Farquhar等[9]和Rai等[11]在无球粒陨石中也发现了富33S的现象,并发现Δ(33S)与Δ(36S)之间并不符合核合成过程以及宇宙射线诱导的碎裂反应过程特征(Δ(36S)/Δ(33S)≈8),推测硫同位素异常来源于太阳前星云的气相反应。同时,Bains-Sahota等[21]、Colman等[22]、Zmolek等[23]通过实验室模拟实验,相继证明了S2F10和CS2的光解和光聚反应可以产生非质量硫同位素分馏效应。他们并用类似于解释氧的非质量同位素分馏来源的分子对称论来解释非质量硫同位素分馏现象的产生,认为是奇质量数同位素(如17O和33S)自旋转动轨道耦合时导致的超精细结构变化能够使这些奇质量数同位素产生非质量同位素效应,但是随后在太古代沉积样品中也发现了Δ(36S)≠0的现象,对这种观点提出了质疑。
(4)2000年,Farquhar等[12]在太古代沉积物样品中检测到非质量硫同位素分馏效应,并通过实验室一系列SO2光分解实验[24],推测190~220 nm深紫外光照射下的SO2光分解反应是产生太古代沉积样品非质量硫同位素分馏效应的过程。随后, Pavlov等[25]通过光化学模型进一步验证了在缺氧大气条件下,在波长λ<220 nm紫外线照射下的SO2光分解反应(SO2→SO+O)是产生非质量硫同位素分馏效应的必要途径。
(5)2003年,在对流层气溶胶中也观测到了明显的非质量硫同位素分馏效应,这表明在现代富氧的大气条件下也能产生非质量硫同位素现象,科学家们对此给予不同解释,Romero等[17]认为是平流层传输给对流层的结果,但是Savarino等[26]认为这可能与激发态SO*2的产生以及随后激发态SO*2与SO2反应(在SO2浓度非常高的条件下)生成SO3和SO有关,SO3随后在现代富氧的条件下迅速转化为H2SO4气溶胶从而使非质量同位素信号得以迁移和保存。他们进一步推论,只要大气中SO2浓度非常高,激发态SO*2与SO2反应可以在220~340 nm范围紫外线照射下发生。
(6)Baroni等[19]提取南极冰芯中近代两次平流层火山事件记录,根据其中记载的非质量硫同位素分馏信息,推测SO3光解反应(SO3→SO2+O)是平流层产生非质量硫同位素分馏效应的途径。
(7)Ohmoto等[27]对Pilbara克拉通岩石研究发现,二氧化硫的气相光化学反应可能不是造成地球硫同位素异常的唯一原因,富含硫的海水与沉积岩中的有机物质之间发生反应也可能产生非质量同位素分馏现象。Watanabe等[28]通过实验室模拟验证了氨基酸还原单质硫、亚硫酸盐和硫酸盐的热化学还原也能产生非质量硫同位素分馏信号。
可见,非质量硫同位素分馏的产生机制仍在探讨过程中,还存在诸多争议,这将随着分馏理论的发展和检测手段的提高得到彻底诠释。
2.1.2 非质量硫同位素的保存
多数科学家认为在缺氧的大气条件下,含硫物种的气相光化学反应可以产生非质量硫同位素分馏信号。支持光化学反应产生机制的科学家们通过实验室模拟和分馏模型,推论非质量硫同位素分馏信号的产生和保存需要3个条件[29]:
(1)缺氧的大气环境的存在,使得紫外线能够穿透对流层,促进了SO2的光分解反应。
(2)需要能够产生多种含硫大气物种的还原性大气条件。在当代含高浓度氧的大气条件下,大气含硫物种迅速被氧化为H2SO4,导致了非质量硫同位素信号的混合稀释。相比较而言,在太古代缺氧大气条件下,大气含硫物种可以多种形式存在,如H2SO4、S8、H2S,从而保存了非质量硫同位素信号。
(3)陆地硫化物氧化风化条件的缺乏。陆地硫化物氧化风化条件可以稀释大气光化学反应所产生的非质量硫同位素信号。
2.2 硫同位素分析技术的建立
硫同位素分析技术的建立,包括所有硫同位素(32S、33S、34S、36S)组成的精确测定,是利用非质量分馏效应深入探讨其来源机制以及拓展其应用的前提。前人在硫同位素分析方法的建立方面已做了较多研究,如采用离子微探针技术[13]、激光微探针技术[14]、气体同位素比质谱(IRMS)技术[30]、聚焦离子束与二次离子质谱仪(SIMS)技术[15]、IRMGC/MS(同位素比气质联用)[31]等现代分析手段进行硫同位素的高精度测量。
微分析技术(如离子微探针技术和激光微探针技术)采用激光束或离子束轰击样品,将得到的待测气体一次引入质谱仪的离子源,可以对固体样品的硫同位素组成进行原位微区(微米级)测定。微分析技术较传统技术具有空间分辨率高、无须破坏样品进行原位测试等优点,但是在离子束或激光束轰击样品时会造成同位素分馏。
对于除微分析技术以外的其他方法,制样技术是关键。传统的制样技术是将硫酸盐转变为SO2,该方法对于δ(34S)的测定具有较好的重现性和精确度,但测定中由于存在氧同位素的干扰,对于δ(33S)的测定精确度偏低;此外,SO2法要求样品含有4 mg以上的硫,这样对一些特殊样品,如月岩样品(每份样品往往只有2~3 mg乃至更低的硫)无法进行分析[32]。因此,不得不寻找新的制样技术。
20世纪60年代以来,一些研究者先后报道了用F2和BrF5作氟化剂制备SF6的方法[33-35]。由于氟为单一同位素,采用SF6作为工作气体进行硫同位素测定,无须对所测的硫化物同位素比值原始数据进行校正,成为硫同位素测定中的主要制样技术。将硫酸盐转变为SF6的方法大体包括酸化和沉淀技术将SO2-4转变为BaSO4、利用还原技术将BaSO4转化为Ag2S、F2、BrF5作氟化剂将Ag2S转变为SF6,然后再经纯化分离,最终将纯SF6导入同位素比质谱仪。近年来,研发出集SO2-4样品富集、样品燃烧、冷冻和转移、解冻和氟化以及SF6工作气体分离和纯化为一体的在线连续流动分析技术[36-37],该技术可用于微克级含硫样品分析,并且大大提高了样品分析效率。
2.3 应用
2.3.1 探索火星大气演化及火星生命痕迹
Farquhar等[10]对火星陨石SNC的几类含硫矿物进行同位素组成分析,发现Nakhal具有明显负的Δ(33S),Lafayette带有较小正Δ(33S)。由于宇宙射线散裂效应只能产生正Δ(33S),因此,结合实验室光化学模拟试验,对非质量硫同位素分馏信号的来源以及迁移进行了推测:火山喷发作用、变质作用或者火星地表富硫物质受热以及光化学分解产生的SO2和H2S在缺氧的火星表面发生气相光化学反应,产生了带有异常同位素组成的含硫物质,随后或者以气溶胶形式沉降到地表,或者与地表物质发生二次反应,将非质量同位素分馏信号以硫酸盐和硫化物的形式保存在火星的地表风化层中。可见,火星陨石非质量硫同位素分馏信号的发现对于追溯火星大气演化、大气与火星地表相互作用、硫在火星大气与地表的循环和迁移具有重要作用。
此外,研究表明34S的分馏可用于寻找火星上的生物活动踪迹。但是大的34S分馏并不能代表一定具有生物活动特征,这是因为火星大气反应(如光化学反应)在带来非质量同位素分馏信号的同时,也会产生34S的分馏。因此,在探寻火星生命活动踪迹时,必须考虑N(33S)/N(32S)和N(36S)/ N(32S)的分馏值。
2.3.2 探索古代大气条件及其演化
2000年,Farquhar等[12]首次在地球早期的沉积岩和变质岩中发现了硫同位素非质量分馏效应,并认为其产生与古代缺氧的大气条件密切相关。研究发现,在年龄大于2 450 Ma沉积物中的硫酸盐和硫化物矿物中,具有明显的非质量同位素效应,Δ(33S)范围为(-2.5~8.2)×10-3;而在年龄小于1.9 Ga的所有硫化物和硫酸盐中Δ(33S)< 0.04×10-3,属于与质量相关的同位素分馏。此后,Farquhar等[12]设计了一系列H2S和SO2的光解实验,高压Xe灯产生波长从200 nm开始的连续光谱。H2S产生的单质硫,其δ(33S)/δ(34S)为0.485±0.005;δ(36S)/δ(34S)为1.86±0.05;SO2产生硫酸,其δ(33S)/δ(34S)为0.649±0.006; δ(36S)/δ(34S)为1.75±0.02。Farquhar由此推论,太古代非质量硫同位素信号的保存可能与空气中缺乏臭氧屏蔽有关,紫外线能够深深穿透平流层,将SO2光解产生带有异常同位素组成的含硫物质S0和H2SO4。此外,S0和H2SO4的同位素组成没有彻底交换,因此它们的部分非质量硫同位素信号被传递到地表。而在现代高O2的大气环境条件下,将SO2或H2S光分解产生的非质量硫同位素分馏信号带到地表是不可能的,因为火山喷发出所有含硫成分都被氧化成硫酸,同位素组成被均一化,非质量分馏特征消失。因此,非质量硫同位素分馏信号可以作为追溯古代大气氧化还原状态的代用指标,它的存在说明当时大气氧浓度很低。Farquhar等推算出当时大气氧浓度是目前大气氧水平的0.015倍。随后,Pavlov等[25]通过模型进一步计算出当时大气氧浓度的上限不足现代大气氧水平的1.0×10-5。
Farquhar的这一发现和推论与古代地质学记录吻合,即认为太古代大气是缺氧的,氧气浓度的急剧增加始于2.4×109a BP。Savarino等[26]随后研究发现,火山喷出的SO2在波长λ<310 nm紫外线照射下发生的光化学反应过程中产生非质量硫同位素分馏效应,而这一波段也正是O3吸收紫外线较强的波段。因而非质量硫同位素分馏效应的测量可用于示踪过去和现代大气层中紫外线穿透O3吸收界面的程度,进而可以作为探索古代大气氧水平进化的一种手段。
还有一种观点认为,地球早期大气成分是氧化性的。Ohmoto等[27]通过对Pilbara克拉通年岩石(曾经的古海洋底部,具有34.6×108a历史)研究发现有赤铁矿存在的标记,他推测这些矿物质很可能是从海底热液喷口涌出的热水与海水中的氧(由光合作用微生物所形成)相互反应的产物。因而他推测古代大气氧的存在有两种可能:一是太古代大气中氧含量存在大的波动,从3.0×109a BP的无氧状态到2.75×109a BP的有氧状态再到2.4× 109a BP的无氧;二是自3.8×109a BP以来大气中持续富氧。他还进一步推测在2.4 Ga沉积岩和变质岩中非质量同位素分馏信号可能来自于富含硫的海水与沉积岩中的有机物质之间发生反应。
可见,非质量硫同位素分馏现象的研究为探索地球早期大气成分及其演化提供了一个新的思路,进而深入系统研究有可能为上述争论画上句号。
2.3.3 探索硫循环
关于地球早期的硫循环存在两种不同的认识。许多学者认为,太古代硫循环与现代硫循环没有太大差别,海洋硫酸盐主要来源于大陆硫化物和硫酸盐的氧化风化[38-41]。还有一种观点认为地球早期是缺氧的,海洋硫酸盐主要来自于火山喷发的含硫物种的光化学氧化。非质量硫同位素分馏效应的发现为认识地球早期的硫来源及其迁移路径开辟了一条独特的新途径,极大地支持了后一种观点。
Farquhar等[16]在南非Orapa钻石硫化物包裹体中也发现了硫的非质量同位素现象,通过对钻石主要成分分析了解到该钻石主要成分为榴辉岩(地幔成分)。非质量硫同位素分馏信号如何从大气传输到地幔?Farquhar等对此作出解释并提出硫循环的概念模型(图2),认为硫循环是从火山喷发的SO2和H2S(Δ(33S)=0)在缺氧的大气条件下发生光化学反应开始的,光化学反应产生一系列不同程度非质量硫同位素的含硫气态分子(其中氧化态的含硫分子带有负的Δ(33S),还原态的含硫分子带有正的Δ(33S)),通过大气沉降作用沉降到地表,其中还原态硫可能在微生物作用下转化为硫化物,这样沉积物中的硫化物具有正的Δ(33S),海洋硫酸盐具有负的Δ(33S),随后通过沉积作用、岩石化、变质作用以及板块俯冲等,带有非质量硫同位素分馏效应的沉积物进入地幔,参与新的循环。这一推论也为探讨大气-地壳--地幔相互作用提供了有力支持。

图2 地球早期硫循环的概念模型Fig.2 Conceptual Model for Primordial Sulfur Cycle in Earth
Ono等[15]在澳大利亚Hamersley盆地的太古代页岩和碳酸岩相硫化物中也发现了明显的非质量硫同位素分馏效应,并对太古代硫循环提出新的见解。研究认为,海洋硫酸盐主要来源于火山喷发含硫气体的光化学氧化,产生的非质量硫同位素信号被化学性质截然不同的气溶胶分别传输:H2SO4(Δ(33S)<0)和自然硫S8(Δ(33S)>0)。这两种气溶胶通过云雨洗脱或大气沉降作用被带入水圈,变成海洋硫酸盐和S8富集区。硫化物中非质量硫同位素分馏信号的保存表明了在低的O2和Fe2+富集的太古代海洋环境中,至少有一些S8共沉淀为黄铁矿,而不是完全被氧化为硫酸盐。因为如果所有的S8被氧化为海洋硫酸盐,那么S8富集区与硫酸盐富集区结合,渐渐转变为硫酸盐富集区,这样非质量硫同位素信号就消除了。
此外,S同位素测量是硫酸盐来源分配指示剂,可作为O同位素测量的补充,对于研究大气中S物种气相和液相氧化路径的相对比例以及追溯其来源具有重要价值。Alexander等[18]测定东南极Vostok冰芯在整个冰期—间冰期循环中的所有硫同位素比值(δ(33S)、δ(34S)、δ(36S)),并结合O同位素测量值得出,S同位素测量是硫酸盐来源分配指示剂,可作为区别对流层与平流层硫酸盐贡献值的一种潜在手段。Δ(33S)和Δ(36S)测量值推导出Vostok冰芯硫酸盐主要来源低纬度海洋生物硫在对流层底部(而非平流层)氧化后搬运至南极的结果。
2.3.4 探寻火山事件对气候的影响
火山活动一直被认为是影响十年到千年尺度上气候变化的重要自然因素。火山喷发,特别是爆炸性的低纬度火山喷发,大量H2S、SO2被喷入到对流层顶部和平流层,在紫外光作用下,与羟基自由基和其他氧化剂反应氧化为硫酸根(SO2-4)或硫酸(H2SO4)气溶胶,后在平流层通过大气环流传输到地球表面,沉积并保存在雪层或火山灰层中。研究发现,在部分冰芯和火山灰的硫酸盐中存在“非质量同位素分馏信号”的踪迹,这种现象是在SO2转化为硫酸根(SO2-4)或硫酸(H2SO4)气溶胶的气相光化学反应过程中产生的。非质量同位素分馏效应研究对于探索与大气化学组成变化及硫的氧化过程相关的细节信息、进一步了解火山事件对气候的影响机制具有重要价值。
Farquhar等[24]通过实验进一步证明,在深紫外光(193 nm)的照射下,SO2的光分解反应可以产生明显的硫同位素非质量分馏。SO2光分解产生的自然硫具有正的Δ(33S)值和相对较低的δ(34S);产生的硫酸具有负的Δ(33S)值和相对较高的δ(34S)。如果冰芯或火山灰样品的Δ(33S)<0,说明这部分硫来自光化学反应形成的硫酸H2SO4;如果冰芯或火山灰样品的Δ(33S)>0,说明这部分硫来自光化学反应形成的元素硫S0。硫酸H2SO4易溶于水,聚集形成气溶胶,因此在空中停留的时间很短,很容易通过云雨洗脱和自然沉降在距离火山活动中心较近的地方沉积下来。而自然硫不溶于水,也难聚合形成气溶胶,因此在空中停留的时间较长,在空中漂移的距离较远,沉降地点远离火山活动中心。因此在火山活动中心附近沉淀的硫化物中的硫主要来自光化学反应产生的硫酸,Δ(33S)为大的负值;远离火山活动中心沉淀的硫化物中的硫主要来自光化学反应产生的元素硫S0,Δ(33S)为较高的正值;位于中间部位的硫化物,Δ(33S)值可能介于两者之间[42]。可见,非质量硫同位素信号可以用于追溯火山事件爆发的位置。
Pavlov等[43]、Baroni等[19]通过冰芯中Agung (1963)和Pinatubo(1991)火山记录的研究表明,只有进入平流层火山(气体)喷发事件才可以产生非质量硫同位素分馏信号。他们认为在现代大气条件下,只有纬度高于20 km平流层中,SO2才可能在190~220 nm紫外光照射下发生光解反应(实验室SO2光解实验中最易产生非质量硫同位素信号的紫外光波段),产生硫的非质量同位素分馏效应,而在纬度低于20 km的对流层和平流层下部,由于缺乏高能量紫外线照射,因而SO2光解反应不能发生,因而不能产生硫的非质量同位素效应。因而,非质量硫同位素信号可以作为区别平流层火山事件和对流层火山事件的依据。
此外,Savarino等[44]研究进一步表明,大型火山事件(几百Tg SO2)和中等火山事件(几十Tg SO2)中SO2的氧化路径不同。Savarino等[44]通过研究南极冰芯火山事件定年分析,揭示了1259年UE(unknown event)火山爆发事件是过去1.0×104a间最大的火山爆发事件,平流层气溶胶释放量(320± 80 Tg)接近Pinatubo火山事件的20倍。但是测定硫酸盐的氧同位素组成发现,1259年UE火山爆发事件的Δ(17O)接近0,而Pinatubo火山事件Δ(17O)介于(0.1~4.7)×10-3之间。结合化学模型推测,大型火山事件喷发的巨量SO2将带有非质量同位素信号的OH“冲出”平流层,随后SO2与O(3P)反应,因而冰芯硫酸盐中并不带有非质量同位素分馏信号。另外,2个火山事件硫同位素组成测定发现,Pinatubo事件中测得正的Δ(33S)和负的Δ(36S)[26];而1259年UE火山爆发事件测得负的Δ(33S)和正的Δ(36S),这也表明不同氧化路径的结果。
3 结语
非质量硫同位素分馏效应的研究是一新领域,受到现有检测技术制约以及对分馏理论认识所限,目前无论是在其来源机制方面,还是在同位素分析方法的建立以及应用领域方面都还处于探索阶段。今后研究的主要趋势:
(1)深入同位素分馏的理论模拟计算研究和模拟实验研究,进一步认识硫的非质量同位素分馏效应的微观来源机制,解决含硫物种参与的气相光化学反应是否是地球物质非质量硫同位素来源的唯一途径这一争论。
(2)硫同位素组成(32S、33S、34S、36S)的高精度测定技术建立是硫同位素研究领域的前沿与趋势。研究在连续流动模式下,开发将气相色谱、燃烧装置、纯化系统集成的在线预处理装置,以SF6为进样气体,是提高硫同位素测定精度的有效途径。此外,解决原位分析技术中可能造成硫同位素分馏这一问题,也是有效简化预处理步骤、得到高精度硫同位素分析数据的重要手段。
(3)从深度和广度上进一步拓展非质量硫同位素分馏在环境和地球科学领域的应用研究。如结合非质量氧同位素分馏研究,并与其他地球化学指标进行对比,提高对于古气候演化信息认识的准确度;加强Δ(33S)、Δ(36S)与火山活动关系的研究,探索与大气化学组成变化及硫的氧化过程相关的细节信息,进一步了解火山事件对气候的影响机制;加大其他行星的非质量硫同位素分馏研究,为太阳系早期大气成分和演化信息提供指导;此外,目前地学界对于古代大气氧水平及其进化、海洋硫酸盐来源及硫循环等地球科学问题仍存在明显不同的认识,这也有赖于非质量同位素分馏理论的进一步完善。
[1] Seal R R.Sulfur Isotope Geochemistry of Sulfide Minerals [J].Reviews in Mineralogy and Geochemistry,2006,61(1): 633-677.
[2] Macnamara J,Thode H G.Comparison of the Isotopic Constitution of Terrestrial and Meteoritic Sulfur[J].Physical Re-view,1950,78(3):307-308.
[3] Hulston J R,Thode H G.Variations in the33S,34S and36S Contents of Meteorites and Their Relation to Chemical and Nuclear Effects[J].Journal of Geophysical Research,1965, 70(14):3475-3484.
[4] Hulston J R.Variations in the Ratios of the Four Stable Sulphur Isotopes in Meteorites and Their Relation to Chemical and Nuclear Effects[D].Hamilton:McMaster University,1964.
[5] Nielsen H.Isotopic Composition of the Major Contributors to Atmospheric Sulfur[J].Tellus,1974,26:213-221.
[6] Ault W V,Jensen M L.Summary of Sulfur Isotope Standards [C]∥Jensen M L.Biogeochemistry of Sulfur Isotopes:NSF Symposium Proceedings.NewHeaven:YaleUniversity, 1963:16-29.
[7] Ding T,Valkiers S,Kipphardt H,et al.Calibrated Sulfur Isotope Abundance Ratios ofThree IAEA Sulfur Isotope Reference Materials and V-CDT with a Reassessment of the Atomic Weight of Sulfur[J].Geochimica et Cosmochimica Acta, 2001,65(15):2433-2437.
[8] Hulston J R,Thode H G.Cosmic-ray-produced36S and33S in the Metallic Phase of Iron Meteorites[J].Journal of Geophysical Research,1965,70(18):4435-4442.
[9] Farquhar J,Jackson T L,Thiemens M H.A33S Enrichment in Ureilite Meteorites:Evidence for a Nebular Sulfur Component[J].Geochimica et Cosmochimica Acta,2000,64(10): 1819-1825.
[10] Farquhar J,Savarino J,Jackson T L,et al.Evidence of Atmospheric Sulphur in the Martian Regolith from Sulphur Isotopes in Meteorites[J].Nature,2000,404:50-52.
[11] Rai V K,Jackson T L,Thiemens M H.Photochemical Massindependent Sulfur Isotopes in Achondritic Meteorites[J]. Science,2005,309:1062-1065.
[12] Farquhar J,Bao H M,Thiemens M H.Atmospheric Influence of Earth's Earliest Sulfur Cycle[J].Science,2000,289:756-758.
[13] Mojzsis S J,Coath C D,Greenwood J P,et al.Mass-independent Isotope Effects in Archean(2.5 to 3.8 Ga)Sedimentary Sulfides DeterminedbyIonMicroprobeAnalysis[J]. Geochimica et Cosmochimica Acta,2003,67(9):1635-1658.
[14] Hu G X,Rumble D,Wang P L.An Ultraviolet Laser Microprobe for the in Situ Analysis of Multisulfur Isotopes and Its Use in Measuring Archean Sulfur Isotope Mass-independent Anomalies[J].Geochimica et Cosmochimica Acta,2003, 67(17):3101-3118.
[15] Ono S H,Eigenbrode J L,Pavlov A A,et al.New Insights into Archean Sulfur Cycle from Mass-independent Sulfur Isotope Records from the Hamersley Basin,Australia[J].Earth and Planetary Science Letters,2003,213(1/2):15-30.
[16] Farquhar J,Wing B A,McKeegan K D,et al.Mass-independent Sulfur of Inclusions in Diamond and Sulfur Recycling on Early Earth[J].Science,2002,298:2369-2372.
[17] Romero A B,Thiemens M H.Mass-independent Sulfur Isotopic Compositions in Present-day Sulfate Aerosols[J].Journal of Geophysical Research,2003,108(D16):4524-4530.
[18] Alexander B,Thiemens M H,Farquhar J,et al.East Antarctic Ice Core Sulfur Isotope Measurements over a Complete Glacial-interglacial Cycle[J].Journal ofGeophysical Research,2003,108(D24):4786-4792.
[19] Baroni M,Thiemens M H,Delmas R J,et al.Mass-independent Sulfur Isotopic Compositions in Stratospheric Volcanic Eruptions[J].Science,2007,315:84-87.
[20] Clayton D D,Ramadurai S.On Presolar Meteoritic Sulphides [J].Nature,1977,265:427-428.
[21] Bains-Sahota S K,Thiemens M H.A Mass-independent Sulfur Isotope Effect in the Nonthermal Formation of S2F10[J]. Journal of Chemical Physics,1989,90(11):6099-6109.
[22] Colman J J,Xu X P,Thiemens M H,et al.Photopolymerization and Mass-independent Sulfur Isotope Fractionations in Carbon Disulfide[J].Science,1996,273:774-776.
[23] Zmolek P,Xu X P,Jackson T,et al.Large Mass Independent Sulfur Isotope Fractionations During the Photopolymerization of12CS2and13CS2[J].Journal of Physical Chemistry,1999, 103(15):2477-2480.
[24] Farquhar J,Savarino J,Airieau S,et al.Observation of Wavelength-sensitiveMass-idependentSulfurIsotopeEffects During SO2Photolysis:Implication for theEarlyEarth Atmosphere[J].Journal of Geophysical Research,2001,106: 32829-32839.
[25] Pavlov A A,Kasting J F.Mass-independent Fractionation of Sulfur Isotopes in Archean Sediments:Strong Evidence for an Anoxic Archean Atmosphere[J].Astrobiology,2002,2(1): 27-41.
[26] Savarino J,Romero A,Cole-Dai J,et al.UV Induced Mass-independent Sulfur Isotope Fractionation in Stratospheric Volcanic Sulfate[J].Geophysical Research Letters,2003,30 (21):2131-2134.
[27] Ohmoto H,Watanabe Y,Ikemi H,et al.Sulphur Isotope Evidence for an Oxic Archaean Atmosphere[J].Nature,2006, 442:908-911.
[28] Watanabe Y,Naraoka H,Ohmoto H.Mass Independent Fractionation of Sulfur Isotopes During Thermochemical Reduction of Native Sulfur,Sulfite and Sulfate by Amino Acids [R].Washington:American Geophysical Union,2007.
[29] Ono S H,Beukes N J,Rumble D,et al.Early Evolution of Atmospheric Oxygen from Multiple-sulfur and Carbon Isotope Records of the 2.9 Ga Mozaan Group of the Pongola Supergroup,Southern Africa[J].South African Journal of Geology, 2006,109(1/2):97-108.
[30] Gao X,Thiemens M H.Systematic Study of Sulfur Isotopic Composition in Iron Meteorites and the Occurrence of Excess33S and36S[J].Geochimica et Cosmochimica Acta,1991, 55(9):2671-2679.
[31] Ono S H,Wing B,Rumble D,et al.High Precision Analysis of All Four Stable Isotopes of Sulfur(32S,33S,34S and36S)at Nanomole Levels Using a Laser Fluorination Isotope-ratiomonitoring Gas Chromatography-mass Spectrometry[J]. Chemical Geology,2006,225(1/2):30-39.
[32] 地质矿产部矿床地质研究所同位素地质研究室.稳定同位素分析方法研究进展[M].北京:北京科学技术出版社,1992.
[33] Puchelt H,Sabels B R,Hoering T C.Preparation of Sulfur Hexafluoride for Isotope Geochemical Analysis[J].Geochimica et Cosmochimica Acta,1971,35(6):625-628.
[34] Thode H G,Rees C E.Measurement of Sulphur Concentrations and the Isotope Ratios33S/32S,34S/32S and36S/32S in Apollo 12 Samples[J].Earth and Planetary Science Letters, 1971,12(4):434-438.
[35] Rees C E.Sulphur Isotope Measurements Using SO2and SF6[J].Geochimica et Cosmochimica Acta,1978,42(4):383-389.
[36] Savarino J,Alexander B,Darmohusodo V,et al.Sulfur and Oxygen Isotope Analysis of Sulfate at Micromole Levels Using a Pyrolysis Technique in a Continuous Flow System[J]. Analytical Chemistry,2001,73(18):4457-4462.
[37] Grassineau N V,Mattey D P,Lowry D.Sulfur Isotope Analysis of Sulfide and Sulfate Minerals by Continuous Flow-isotope Ratio Mass Spectrometry[J].Analytical Chemistry, 2001,73(2):220-225.
[38] Ohmoto H,Kakegawa T,Lowe D R.3.4-billion-year-old Biogenic Pyrites from Barberton,South Africa:Sulfur Isotope Evidence[J].Science,1993,262:555-557.
[39] Ohmoto H.Archaean Atmosphere,Hydrosphere and Biosphere[C]∥Eriksson P G,Altermann W,Nelson D R,et al. The Precambrian Earth:Tempos and Events.Amsterdam: Elsevier,2004:361-388.
[40] Huston D L,Logan G A.Barite,BIFs and Bugs:Evidence for the Evolution of the Earth's Early Hydrosphere[J].Earth and Planetary Science Letters,2004,220(1/2):41-55.
[41] Kakegawa T,Ohmoto H.Sulfur Isotope Evidence for the Origin of 3.4 to 3.1 Ga Pyrite at the Princeton Gold Mine,Barberton Greenstone Belt,South Africa[J].Precambrian Research,1999,96(3):209-224.
[42] 侯可军,李延河,万德芳.鞍山—本溪地区条带状硅铁建造的硫同位素非质量分馏对太古代大气氧水平和硫循环的制约[J].中国科学(D辑),2007,37(8):997-1003.
[43] Pavlov A A,Mills M J,Toon O B.Mystery of the Volcanic Mass-independent Sulfur Isotope Fractionation Signature in the Antarctic Ice Core[J].Geophysical Research Letters, 2005,32:L12816/1-L12816/4.
[44] Savarino J,Bekki S,Cole-Dai J,et al.Evidence from Sulfate Mass Independent Oxygen Isotopic Compositions of Dramatic Changes in Atmospheric Oxidation Following Massive Volcanic Eruptions[J].Journal of Geophysical Research,2003, 108(D21):4671-4676.
Overview of Mass Independent Sulfur Isotope Fractionation
MA Hong-mei,LI Yuan-sheng,J IANG Su,AN Chun-lei
(Key Laboratory of Polar Science of State Oceanic A dministration,Polar Research Institute of China,Shanghai200136,China)
The research on mass independent sulfur isotope fractionation is one of the advancing fronts in stable isotope geochemistry.Based on brief introduction of related theory,the recent progress in mass independent sulfur isotope fractionation is summarized.The micro origin mechanism of mass independent sulfur isotope fractionation is still controversial;taking SF6as the working gas is the main preparation technology for high precise measurement of sulfur isotope composition;the study on mass independent sulfur isotope fractionation provides new approach to explain many important geologic events,which include atmospheric evolvement and possible biological activity exploration of Mars,primordial atmospheric oxidizing condition,primordial sulfur cycle of earth,effect of volcanic activity to climate,and so on.Finally,the further research trend of mass independent sulfur isotope fractionation is discussed.
sulfur isotope;fractionation;origin mechanism;preparation technology;geology;research trend
P597+.2
A
1672-6561(2010)02-0176-07
2009-07-08
国家自然科学基金项目(40806074);国家科技支撑计划项目(2006BAB18B01)
马红梅(1976-),女,安徽霍山人,助理研究员,工学博士,从事冰川化学和气候记录研究。E-mail:mahongmei@pric.gov.cn
