氮铁共掺锐钛矿相TiO2电子结构和光学性质的第一性原理研究*
2010-09-08张学军高攀柳清菊
张学军高攀柳清菊
1)(云南大学物理科学技术学院,云南省高校纳米材料与技术重点实验室,昆明650091)
2)(湖南城市学院物理与电信工程系,益阳413000)
(2009年10月13日收到;2009年11月15日收到修改稿)
氮铁共掺锐钛矿相TiO2电子结构和光学性质的第一性原理研究*
张学军1)2)†高攀1)柳清菊1)
1)(云南大学物理科学技术学院,云南省高校纳米材料与技术重点实验室,昆明650091)
2)(湖南城市学院物理与电信工程系,益阳413000)
(2009年10月13日收到;2009年11月15日收到修改稿)
本文采用基于密度泛函理论的平面波超软赝势方法研究了N,Fe共掺杂TiO2的晶体结构、电子结构和光学性质.研究表明,N,Fe共掺杂TiO2的晶格体积、原子间的键长及原子的电荷量发生变化,导致晶体中产生八面体偶极矩,并因此光生电子-空穴对有效分离,提高TiO2的光催化活性;N,Fe共掺杂同时在导带底和价带顶形成了杂质能级,使TiO2的禁带宽度变窄,光吸收带边红移到可见光区,这些杂质能级可以降低光生载流子的复合概率,提高TiO2的光催化效率;与Fe掺杂TiO2的态密度相比,共掺杂位于价带顶的杂质能级的态密度峰明显增大,导致电子从杂质能级跃迁到导带的概率增加,使其对太阳能的利用率提高;在不考虑杂质能级的情况下,与纯TiO2相比,N,Fe共掺杂TiO2的带边位置只有微小变化,因此N,Fe共掺杂TiO2的强氧化还原能力得以保持.
第一性原理计算,氮铁共掺杂锐钛矿相TiO2,电子结构,光学性质
PACC:7115M,7115H,7115A
1. 引言
近年来,锐钛矿相TiO2由于具有良好的光催化性能而得到了广泛的研究及初步应用[1—15].然而,纯的锐钛矿相TiO2具有较宽的禁带宽度(约3.23 eV),只能在占太阳光能量不足5%的紫外光照射下才显示出光催化活性,对太阳能利用率很低;同时由于其被光激发产生的电子-空穴对的复合概率较高,导致其光量子效率低.上述因素限制了TiO2在光催化领域的应用,必须对其进行改性.
实验研究发现,过渡金属离子与N离子掺杂能有效改变其光吸收特性和减少光生电子和空穴的复合.Choi等[16]研究了21种金属离子掺杂TiO2的光催化效果,以CHCl3氧化和CCl4还原为模型反应.其结果表明,掺杂0.1%—0.5%金属离子的TiO2以Fe3+的效果最好.普遍认为,Fe3+掺杂改性TiO2的机理是通过Fe3+取代TiO2晶格中的Ti4+,在晶格中形成了浅电荷俘获陷阱,减少了电子-空穴对的复合,从而提高光催化效率[17—19].Asahi等[20]报道了非金属元素C,N,F,P,S等取代TiO2晶体中少量的晶格O,能使TiO2的禁带窄化,扩大TiO2的光响应范围.
最近研究[21—28]表明,对TiO2有选择性地进行不同离子的共掺杂,可以利用离子间的协同作用进一步提高TiO2光催化剂的光吸收范围和光催化活性,得到的光催化剂具有比单一元素掺杂更高的光催化性能.黄东升等[29]认为N,Fe3+共掺杂的TiO2中存在N和Fe3+的协同作用:N掺杂减小了TiO2的带隙宽度,增强了其对可见光的响应,而Fe3+掺杂抑制了光生载流子的复合,提高了反应效率.Yang等[30]制备的C,S,N,Fe3+共掺杂TiO2光催化剂在可见光区有很大的光吸收系数,其光催化性能优于C,S,N和Fe3+分别单掺杂TiO2,认为Fe3+掺杂作为浅俘获载流子(光生电子或者空穴)的陷阱,分开了光生电子和空穴到达晶体表面的时间,从而提高了光催化效率;C,S,N和Fe3+掺杂在TiO2的带隙中引入了新的杂质能级,这些杂质能级使TiO2的带隙宽度变窄,能提高可见光区的吸收.
过渡金属与N离子掺杂改性TiO2在实验研究方面已经取得了一定的进展,但同时也存在不少问题,其中缺乏对实验结果的机理分析,离子间的协同作用机理还不明确.与实验研究相比,在计算机模拟基础上进行的理论计算分析可克服实验因素的影响,能够突出离子掺杂效应中的主要因素,并且可以分析离子掺杂引起的TiO2微观结构的变化,因此更有利于研究掺杂对TiO2光催化性能的影响机理.为此,本文采用基于密度泛函理论的第一性原理平面波超软赝势方法计算并分析比较了N,Fe共掺杂TiO2与N掺杂TiO2,Fe掺杂TiO2及纯TiO2在晶体结构、电子结构、光学性质等方面的差异,从理论上解释了N,Fe共掺杂对TiO2光催化性能影响的机理.
2. 计算模型和计算方法
2.1. 计算模型
锐钛矿相TiO2的晶体结构属四方晶系,空间群为I41/amd,点群为D194h,一个正格矢晶胞中含有4个Ti原子、8个O原子,如图1(a)所示.本文采用的N,Fe共掺杂超晶胞模型是由两个正格矢晶胞分别沿a轴和b轴排列而成,如图1(b)所示,其中一个Ti原子被一个Fe原子所取代,一个O原子被一个N原子所取代,因此一个超晶胞中就含有48个原子:15个Ti原子、31个O原子、1个Fe原子和1个N原子,掺杂原子的摩尔浓度为2.08%.其他的对比模型:N掺杂TiO2超晶胞、Fe掺杂TiO2超晶胞以及纯TiO2超晶胞按照同样的方法进行构建.

图1 锐钛矿相TiO2的正格矢晶胞和N,Fe共掺杂超晶胞模型(a)锐钛矿相TiO2的正格矢晶胞,(b)N,Fe共掺杂锐钛矿相TiO2的2×2×1超晶胞及掺杂原子的位置
2.2. 计算方法
本文所有计算工作采用基于密度泛函理论的平面波超软赝势方法进行,应用Accelrys公司开发的Materials Studio 4.1中的CASTEP模块进行计算.CASTEP是基于密度泛函理论的从头算量子力学程序,利用总能量平面波赝势方法,将离子势用赝势代替,电子波函数通过平面波基组展开,电子-电子相互作用的交换关联能由局域密度近似(local density approximation,简记为LDA)或广义梯度近似(generalized gradient approximation,简记为GGA)进行校正,是目前较为准确的电子结构计算的理论方法[31,32].价电子平面波函数的截断能设置为380 eV,交换关联能应用LDA中的CA-PZ函数[33,34],所有的计算均在倒易空间中进行,这样可以同时提高计算的效率和精度.对不可约Brillouin区的积分计算采用3×7×3的Monkorst-pack特殊k点进行取样求和,快速Fourier变换的网格设置为45×45× 54,迭代过程中的收敛标准设置为:原子位移不大于5×10-4nm,原子间作用力不大于0.01 eV/nm,原子间的内应力不大于0.02 GPa,体系总能量的变化每原子不大于5×10-6eV.为了得到稳定精确的计算结果,先根据能量最小化原理得到合适的晶格常数,并优化其内坐标,然后在此基础上进行电子结构和光学性质的计算.在光学性质的计算中采用非极化多晶模型,并使用“剪刀算符”对结果进行修正,以便于与实验数据进行比较[35,36].
3. 计算结果与讨论
3.1. 杂质形成能及晶体结构
杂质形成能(Ef)通常被用作分析比较不同离子掺杂的相对难易程度.本文Ef定义为[37]
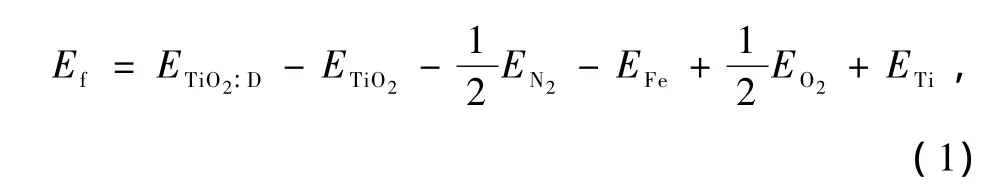
其中ETiO2:D是掺杂后的体系总能量,ETiO2是与掺杂体系相同大小的纯TiO2的超晶胞体系总能量,EN2,EO2,EFe,ETi分别是N2分子、O2分子、单质Fe、单质Ti的能量.所得到的结果如表1所示.从表1中可以看出,N,Fe共掺杂TiO2的杂质形成能比N掺杂TiO2,Fe掺杂TiO2的大,表明合成N,Fe共掺杂TiO2需要较大的能量.
Sato等[38]报道了由晶体内的八面体偶极矩产生的内部局域电场有利于光生电子-空穴对的分离,从而改善光催化剂的光催化活性.表1中同时列出了掺杂体系结构优化后的晶格畸变、平均键长、由Mulliken布居分析得到的平均净电荷以及八面体的平均偶极矩.从表1中可以看出,掺杂后Ti—O键长的变化很小.在Fe掺杂TiO2体系中Fe—O键长明显小于Ti—O键长,同时由于掺入晶体的Fe3+半径64 pm略小于Ti4+半径68 pm,使其晶格体积大幅减小;而N掺杂TiO2后,晶体中的Ti—O键长和Ti—N键长都比纯TiO2的键长长,同时由于掺入晶体的N3-半径171 pm略大于O2-半径140 pm,使其晶格体积趋于增大;当N,Fe共掺杂TiO2后,晶体中的Ti—O键长和Ti—N键长以及Fe—O键长都比纯TiO2的键长短,故晶格体积仍减小较多.由于掺杂后晶格发生了畸变,原子间的键长及原子的电荷量都发生了变化,这意味着掺杂后TiO6八面体和以Fe离子为中心的FeO6八面体中负电荷的中心不再与正电荷的中心重合,从而产生内部偶极矩[35,36].而N,Fe共掺杂的偶极矩变化尤其明显,这说明N,Fe共掺杂能使TiO2的光生电子-空穴对更有效分离,降低其复合概率,将会更有效地提高TiO2的光催化活性.
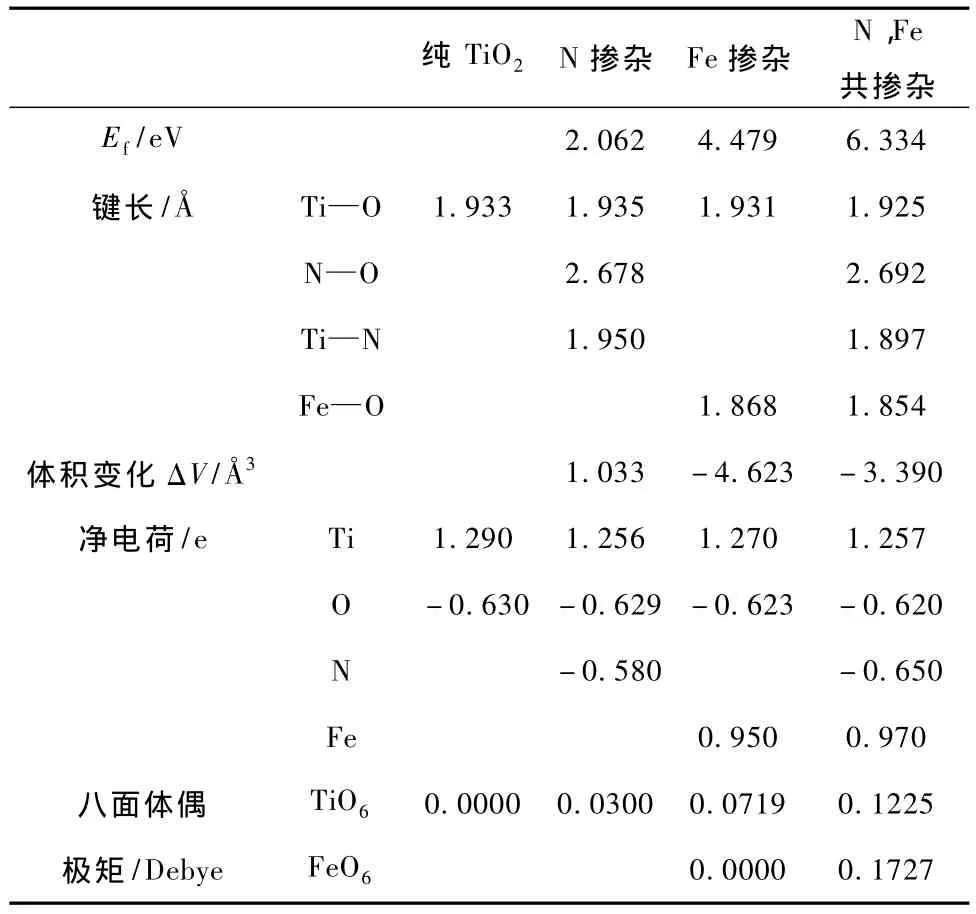
表1 掺杂体系结构优化后的物理参数
3.2. 电子结构的计算结果
计算得到的各体系的能带结构如图2所示.可以看到,由于掺杂后晶体对称性的下降,从而使能级的简并度降低、发生分裂.在不考虑杂质能级的情况下,N,Fe分别单掺杂及两者共掺杂后TiO2的禁带宽度都变宽,分别由纯TiO2的2.54 eV变为2.56,2.76和2.60 eV.对于N掺杂TiO2,在价带上方形成了三条杂质能级,这些杂质能级与价带顶(VBM)充分交叠.而对于Fe掺杂TiO2,则在导带下方形成了两条杂质能级,它们与导带底(CBM)充分交叠,同时还在Fermi能级附近形成了3条杂质能级(张勇等[39]定义这些能级为局域能级).对于N,Fe共同掺杂TiO2,则同时在价带上方与导带下方形成了杂质能级,由于晶体的对称性进一步下降,在价带上方与导带下方的杂质能级相对相应的单质掺杂而言有较明显的分离;在Fermi能级附近形成的3条局域能级与价带上方的杂质能级充分交叠,而且这3条局域能级更集中.可见,金属离子与非金属离子掺入TiO2都会改变TiO2的电子结构,即形成新的杂质能级,这些杂质能级对实现可见光响应型掺杂TiO2具有重要作用.
为了进一步比较不同离子掺杂对电子结构的影响,图3给出了计算得到的总态密度图和禁带附近的分波态密度图.从图3(a)中可以看到,相对纯TiO2而言,N,Fe单掺杂以及两者共掺杂后TiO2的价带带边能级和导带带边能级及带隙宽度都发生了不同程度的变化.

图2 计算得到的能带结构图Eg为带隙.(a)纯TiO2,(b)N掺杂TiO2,(c)Fe掺杂TiO2,(d)N,Fe共掺杂TiO2
由图3(b)可以看出,在纯TiO2中价带和导带由O原子的2p轨道和Ti原子的3d轨道组成,其中价带主要是O原子的2p轨道的贡献,导带主要是Ti原子的3d轨道的贡献.在纯TiO2晶胞中一个Ti4+被6个O2-包围,构成TiO6八面体,根据晶体场理论,Ti原子的3d轨道分裂为两组:能量较高的eg(d2z,dx2-y2)轨道和能量较低的t2g(dxy,dyz,dxz)轨道,这就使TiO2的导带分为两部分,其中导带上部分由O原子的2p轨道和Ti原子的eg轨道构成,导带下部分由O原子的2p轨道和Ti原子的t2g轨道构成.图3(c)—(e)分别显示出N,Fe单掺杂及两者共掺杂后在TiO2的导带与价带之间的不同位置形成了新的杂质能级,分别由N原子的2p轨道或者Fe原子的3d轨道与O原子的2p轨道、Ti原子的3d轨道杂化形成,这种杂化效果有利于光生电子和空穴的迁移,也有利于光催化反应的进行[40].由于在禁带中形成了杂质能级,使价带中的电子只需吸收能量较小的光子就可以跃迁到杂质能级中,然后再次吸收光子而跃迁到导带中,从而能使TiO2的光吸收范围拓展到可见光区.

图3 数值计算得到的各种离子掺杂的态密度EF为Fermi能级.(a)总态密度的比较,(b)—(e)为各种离子掺杂在禁带附近的分波态密度图,(b)纯TiO2,(c)N掺杂TiO2,(d)Fe掺杂TiO2,(e)N,Fe共掺杂TiO2
比较图3(c)—(e)可以看出,N,Fe单掺及两者共掺三种方式在细节上有一定差别:1)N掺杂进入TiO2晶格中,在晶体中形成O—Ti—N键,导致能带结构发生变化:价带由O原子的2p轨道、Ti原子的3d轨道和N原子的2p轨道组成,在价带上方位于N原子的2p轨道上态密度有两个波峰,N原子的2p轨道与O原子的2p轨道.Ti原子的3d轨道杂化形成了3条杂质能级,主要是N原子的2p轨道的贡献.这些杂质能级位于Fermi能级以下,被电子占据,且与TiO2的价带顶充分交叠,可以成为光生空穴的有效俘获陷阱[41],同时引起TiO2的价带宽度增加.N掺杂TiO2的导带由O原子的2p轨道、Ti原子的3d轨道和N原子的2p轨道组成,由于在导带区位于N原子的2p轨道上的态密度极小,因此导带主要是Ti原子的3d轨道、O原子的2p轨道的贡献,N原子掺杂对导带只有很小的影响,导带带边能级略微下移.从而使TiO2的禁带宽度变窄,考虑杂质能级其带隙宽度减小为1.95 eV.2)Fe掺杂TiO2的价带主要由O原子的2p轨道组成,导带由Ti原子的3d轨道、O原子的2p轨道和Fe原子的3d轨道组成.Fe原子的3d轨道在晶体场中也分裂为eg轨道和t2g轨道.在导带底附近位于Fe原子的eg轨道上态密度有一个波峰,峰形平滑,峰值较小,Fe原子的eg轨道与Ti原子的的t2g轨道、O原子的2p轨道杂化在TiO2的导带下方形成了两条杂质能级,主要是Fe原子的eg轨道的贡献,这些杂质能级位于Fermi能级之上,并没有被电子占据,且与TiO2的导带底充分交叠,这些杂质能级可以成为光生电子的有效俘获陷阱[17—19].在Fermi能级附近位于Fe原子的t2g轨道上态密度有一个波峰,峰形尖,峰值大,Fe原子的t2g轨道与O原子的2p轨道、Ti原子的3d轨道杂化形成了3条局域能级,主要是Fe原子的t2g轨道的贡献,Fermi能级穿过这些杂质能级,意味着在Fermi能级以下的杂质能级也能被电子所占据,这些电子只需吸收较小的能量就能跃迁到导带上,能极大地影响光吸收特性,使其对可见光的利用率提高.但是这些杂质能级属于深掺杂能级,如果Fe掺杂浓度过高,会成为光生电子-空穴的复合中心,反而降低光量子产率,从而使TiO2的光催化性能大大减弱[17,18].在Fermi能级附近主要由Fe原子的t2g轨道形成的局域能级在TiO2的光谱响应中扮演了重要角色[39],考虑杂质能级其带隙宽度减小为2.52 eV.3)N,Fe共掺杂TiO2同时在导带下方与价带上方形成了新的杂质能级,由于晶体对称性进一步下降,各杂质能级相对单掺杂而言有较明显的分离,考虑杂质能级其带隙宽度减小为1.68 eV,导致共掺杂TiO2的光吸收带边红移到可见光区,并且这些杂质能级可以成为俘获光生载流子(电子或者空穴)的陷阱,有利于光生电子-空穴对的进一步分离,从而有效地提高了TiO2的光催化性能.在导带下方,由于Fe原子的eg轨道与Ti原子的t2g轨道、O原子的2p轨道杂化,位于Fe原子的eg轨道上态密度有两个波峰,说明Fe原子的eg轨道更加离散,也就是说在导带下方主要由Fe原子的eg轨道形成的两条杂质能级更加分裂;在Fermi能级附近形成了3条杂质能级,主要是Fe原子的t2g轨道的贡献,这三条杂质能级与价带上方主要由N原子的2p轨道形成的杂质能级充分交叠,主要原因是: N,Fe共掺TiO2以后,在Fermi能级附近N原子的2p轨道上的电子与Fe原子的t2g轨道上的电子发生强烈关联相互作用[42,43],致使Fe原子的t2g轨道上的电子向N原子的2p轨道发生了移动,从而Fermi能级附近杂质能级的态密度的峰值增大,峰形变尖,Fermi能级附近的局域能级与价带上方主要由N原子的2p轨道形成的杂质能级发生交叠,而且更加集中.这些局域能级可以成为光生空穴的有效俘获陷阱,促进了光生载流子在晶体内的扩散过程,延长了光生载流子的寿命,抑制了光生电子-空穴对的复合,提高了反应效率[29].由于N原子的2p轨道、Fe原子的3d轨道与Ti原子的3d轨道、O原子的2p轨道杂化,与Fe单掺杂TiO2的态密度相比,N,Fe共掺杂位于价带顶的杂质能级的态密度峰明显增大,导致电子从杂质能级跃迁到导带的概率增加,从而使其对可见光的吸收率提高[30].也就是说N,Fe共掺杂TiO2产生的协同作用将共同提高TiO2在可见光区的光催化活性.
3.3. 光学性质的计算结果
在电子结构的计算基础上,本文采用非极化多晶模型,并由“剪刀算符”进行修正,计算了各种离子掺杂的光吸收谱图,结果如图4所示.在计算中,根据带隙的理论计算值2.54 eV与实验值3.23 eV之间的差值,剪刀算符设置为0.69 eV[35,36,41].从图4中可以看出,经过N,Fe分别单掺及两者共掺杂后,TiO2表现出不同程度的吸收阈值波长的红移,并具有以下特点:1)纯TiO2的吸收带边位于380nm附近,而且吸收带边并不是垂直变化的,有一定的弯曲,这些计算结果与实验结果[44]符合得很好,因此在计算光学性质时使用剪刀算符修正是必要的;2)N掺杂后,吸收带边大约位于470nm,这与Asahi等[20]的实验结果基本一致;3)Fe掺杂后,吸收带边大约位于390nm,这与文献[45,46]报道的实验结果基本相符;4)N,Fe共掺后,吸收带边大约位于520nm,这与文献[29]报道的实验结果基本一致.可见共掺后TiO2的红移最大.

图4 计算得到的各种离子掺杂TiO2的光吸收谱图
图4还表明,不同离子掺杂以后,TiO2在可见光区的吸收系数均增大,而N,Fe共掺杂TiO2的增加最明显,而且在紫外光区的光吸收系数也有明显地提高.也就是说,N,Fe共掺杂使TiO2在紫外光区和可见光区有了更强的光吸收特性,产生这一现象的原因可能是:1)由于N原子的2p轨道、Fe原子的3d轨道与Ti原子的3d轨道、O原子的2p轨道杂化,在TiO2的导带与价带之间形成了新的杂质能级,电子可以从价带上方的杂质能级跃迁到TiO2的导带,电子也可以从价带上方的杂质能级跃迁导带底附近的杂质能级,电子还可以从价带跃迁导带底附近的杂质能级[30],还有可能价带中的电子只需吸收能量较小的光子跃迁到杂质能级中,然后再次吸收光子而跃迁到导带中,从而提高了TiO2在可见光区的吸收系数;2)位于价带顶的杂质能级的态密度峰值明显变大,即电子占据概率增大,从而电子从价带或者价带上方杂质能级跃迁到导带下方的杂质能级或者导带的概率增加,使其对紫外光和可见光的吸收率明显提高.
3.4. 带边位置的计算结果
对于光生电子-空穴来说,电荷迁移的速率和概率及其光催化反应的能力,取决于半导体的导带和价带边的位置及吸附物种的氧化还原电位[47].热力学允许的光催化氧化-还原反应,要求受体电势比半导体导带带边电势低(更正),给体电势比半导体价带带边电势高(更负),这样半导体被激发产生的光生电子或者空穴才能传给基态的吸附分子.为了进一步研究离子掺杂后TiO2光催化性能的改变,本文采用如下公式来计算体系的带边位置(即相对于标准氢电极的氧化还原电势)[40,48]:

式中ECB,EVB分别是导带带边的还原电势和价带带边的氧化电势(相对标准H电极),X是组成半导体体系各原子电负性的几何平均值,Ee是自由电子以H为标准时的电势(~4.5 eV),Eg采用的是经过剪刀算符修正的体系的带隙值.按照(2)式计算纯TiO2的导带带边位置为-0.303 eV,TiO2的价带带边位置为2.927 eV,这个结果与文献[47]报道的实验数据非常接近.根据(2)式计算得到了不同离子掺杂TiO2的带边位置及其光波吸收阈值,如图5所示.图5中竖直轴指示半导体体系氧化还原势大小,ENHE/eV是指半导体体系的带边相对标准H电极(normal hydrogen electrode,简记为NHE)的电势,图中粗短实线代表在不考虑杂质能级的情况下N,Fe单掺杂及两者共掺杂TiO2光催化剂的带边位置;在考虑杂质能级的情况下,N掺杂TiO2光催化剂的导带带边位置及Fe掺杂TiO2光催化剂的价带带边位置也用粗短实线表示;粗短点划线代表在考虑杂质能级的情况下N掺杂及N,Fe共掺杂TiO2光催化剂的价带带边位置,粗短虚线代表在考虑杂质能级的情况下Fe掺杂及N,Fe共掺杂TiO2光催化剂的导带带边位置.从图5可以看出,在不考虑杂质能级的情况下,N,Fe单掺杂及两者共掺杂TiO2光催化剂与纯TiO2相比较,掺杂后的导带带边位置稍微向上移(即还原电势更负),而价带带边位置则稍微向下移(即氧化电势更正),只是带边位置的改变是极其微小的.众所周知,H2O2及O3是很强的氧化剂,它们的氧化电势(相对标准H电极)分别是1.77 eV,2.07 eV.这意味着N,Fe单掺杂及两者共掺TiO2光催化剂具有很强的氧化还原能力.又由于N,Fe共掺杂在TiO2的价带上方和导带下方都形成了新的杂质能级,带隙宽度变窄,使N,Fe共掺杂TiO2光催化剂可以吸收更多可见光,并且使光生电子-空穴得到了更有效的分离,从而实现了共掺杂TiO2在可见光激发下具有很强的光催化活性.
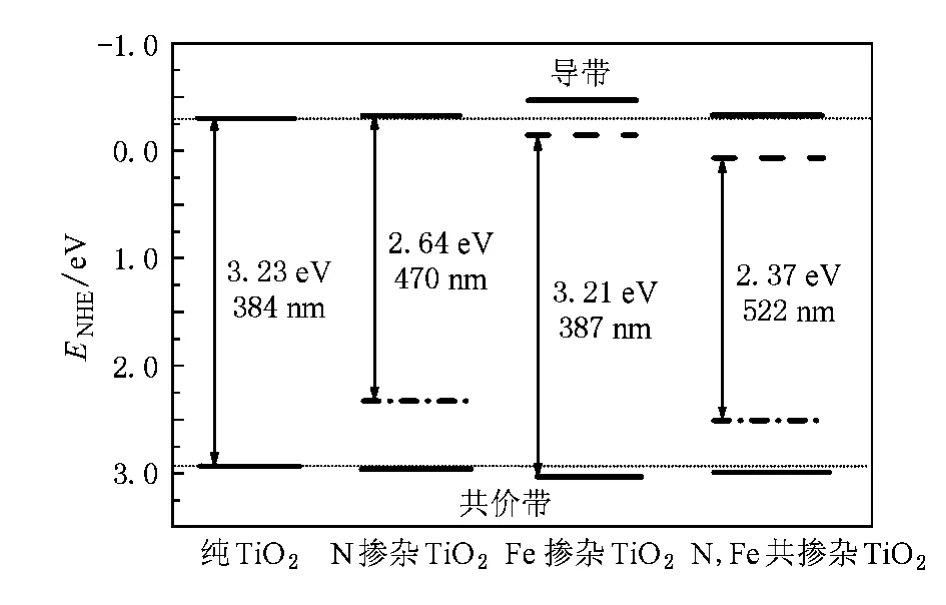
图5 计算得到的不同离子掺杂TiO2的带边位置及其光谱吸收阈值能级之间的距离是被剪刀算符修正过的
4. 结论
本文采用第一性原理计算了N,Fe共掺对于TiO2的晶体结构、电子结构和光学性质的影响,研究了N,Fe共掺杂TiO2对其光催化活性的影响机理以及离子间的协同作用机理.计算结果表明:掺杂后TiO2晶格发生畸变、原子间的键长及原子的电荷量也发生了变化,导致体系中的八面体偶极矩增大,从而有利于光生电子-空穴对的分离,将能有效提高TiO2的光催化活性;N,Fe共掺杂后,由于N原子的2p轨道、Fe原子的3d轨道与Ti原子的3d轨道、O原子的2p轨道杂化,在TiO2的导带与价带之间形成了新的杂质能级,这些杂质能级一方面减小了TiO2的带隙宽度,使光吸收曲线红移到可见光区,另一方面有利于光生电子-空穴对的分离,提高了TiO2的光催化效率;Fermi能级附近的杂质能级主要是Fe的t2g轨道的贡献,Fermi能级穿过杂质能级能极大地影响光吸收特性,使其对可见光的利用率提高,但是如果Fe掺杂浓度过高,则会成为光生电子-空穴的复合中心,反而降低光量子产率;N,Fe共掺杂使位于TiO2价带顶的杂质能级的态密度峰明显增大,导致电子从杂质能级跃迁到导带的概率增加,使其对太阳能的利用率提高;另外,TiO2的带边位置经共掺杂以后只有微小的变化,N,Fe共掺杂TiO2仍然具有强氧化还原能力,因此既可实现TiO2对可见光的响应,同时又保持了原有的紫外光下的强的催化活性.综合分析比较计算结果,N,Fe共掺杂TiO2的光催化活性将比N,Fe单掺杂TiO2的更好,本文计算预测的结果与相应文献[29]的实验结果一致.
感谢云南大学高性能计算中心在数值计算方面提供的技术支持与帮助.
[1]Kanisaka H,A dachi T,Yamashita K 2005 J.Chem.Phys.123 84704
[2]Chen X,Mao S 2007 Chem.Rev.107 2891
[3]Khan S U M,Al-Shahry M,Ingler W B 2002 Science 297 2243
[4]Yu J X,Fu M,Ji G F,Chen X R 2009 Chin.Phys.B 18 269
[5]Zhu J,Yu J X,Wang Y J,Chen X R,Jing F Q 2008 Chin. Phys.B 17 2216
[6]Hou Q Y,Zhang Y,Zhang T 2008 Acta Phys.Sin.57 1862(in Chinese)[侯清玉、张跃、张涛2008物理学报57 1862]
[7]Lin F,Zheng F W,Ouyang F P 2009 Acta Phys.Sin.58 193 (in Chinese)[林峰、郑法伟、欧阳方平2009物理学报58 193]
[8]Sun H W,Zhang X J,Zhang Z Y,Chen Y S,Crittenden J C 2009 Environ.Pollut.157 1165
[9]Chen F,Zou W W,Qu W W,Zhang J L 2009 Catal.Commun. 10 1510
[10]Ananpattarachai J,Kajitvichyanukul P,Seraphin S 2009 J. Hazard Mater.168 253
[11]Park Y,Kim W,Park H,Tachikawa T,Majima T,Choi W 2009 Appl.Catal.B 191 355
[12]Yu H Z,Peng J B,Liu J C 2009 Acta Phys.Sin.58 669(in Chinese)[於黄忠、彭俊彪、刘金成2009物理学报58 669]
[13]Hou Q Y,Zhang Y,Zhang T 2008 Acta Phys.Sin.57 3155(in Chinese)[侯清玉、张跃、张涛2008物理学报57 3155]
[14]Liang L Y,Dai S Y,Fang X Q,Hu L H 2008 Acta Phys.Sin. 57 1956(in Chinese)[梁林云、戴松元、方霞琴、胡林华2008物理学报57 1956]
[15]Ma X G,Jiang J J,Liang P 2008 Acta Phys.Sin.57 3120(in Chinese)[马新国、江建军、梁培2008物理学报57 3120]
[16]Choi W,Termin A,Hoffmann M R 1994 J.Phys.Chem.98 13669
[17]Wang C,Bahnemmannt D,Dohrmann J 2000 Chem.Commun. 16 1539
[18]Wang C,Li Q,Wang R 2004 J.Mater.Sci.39 1899
[19]Zhang Z,Wang C,Zakaria R,Ying J 1998 J.Phys.Chem.B 102 10871
[20]Asahi R,Morikawa T,Ohwaki T,Aoki O K,Taga Y 2001 Science 293 269
[21]Luo H,Takata T,Lee Y,Zhao J,Domen K,Yan Y 2004 Chem. Mater.16 846
[22]Shi J W,Zheng J T,Hu Y,Zhao Y C 2007 Mater.Chem. Phys.106 247
[23]Liu H Y,Gao L 2004 Chem.Lett.33 730
[24]Chen Q L,Tang C Q 2009 Acta Phys.Chim.Sin.25 915(in Chinese)[陈琦丽、唐超群2009物理化学学报25 915]
[25]Xie Y,Li Y Z,Zhao X J 2007 J.Mole Cata.A 277 119
[26]Gu D E,Yang B C,Hu Y D 2008 Cataly.Commun.9 1472
[27]Huang L H,Sun C,Liu Y L 2007 Appl.Surf.Sci.253 7029
[28]Pelaez M,Cruz A,Stathato S,Falaras P,Dionysiou D 2009 Catal.Today 144 19
[29]Huang D S,Chen C F,Li Y H,Zeng R J 2007 Chin.J.Inorg. Chem.23 728(in Chinese)[黄东升、陈朝凤、李玉花、曾人杰2007无机化学学报23 728]
[30]Yang X X,Cao C D,Erickson L,Hohn K,Maghirang R,Klabunde K 2009 Appl.Cataly.B 91 657
[31]Segall M D,Lindan P J D,Probert M J,Pickard C J,Hasnip P J,Clark S J,Payne M C 2002 J.Phys.Cond.Matt.14 2717
[32]Keiji W,Masatoshi S,Hideaki T 2001 Electrochemistry 69 407
[33]Ceperley D M,Alder B J 1980 Phys.Rev.Lett.45 566
[34]Perdew J P,Zunger A 1981 Phy.Rev.B 23 5048
[35]Zhao Z Y,Liu Q J,Zhu Z Q,Zhang J 2008 Acta Phys.Sin.57 3760(in Chinese)[赵宗彦、柳清菊、朱忠其、张瑾2008物理学报57 3760]
[36]Zhao Z Y,Liu Q J,Zhu Z Q,Zhang J,Liu Q 2008 J.Funct. Mater.39 953(in Chinese)[赵宗彦、柳清菊、朱忠其、张瑾、刘强2008功能材料39 953]
[37]Cui X Y,Medvedeva J E,Delley B,Freeman A J,Newman N,Stampfl C 2005 Phys.Rev.Lett.95 256404
[38]Sato J,Kobayashi H,Inoue Y 2003 J.Phys.Chem.B 107 7970
[39]Zhang Y,Tang C Q,Dai J 2005 Acta Phys.Sin.54 323(in Chinese)[张勇、唐超群、戴君2005物理学报54 323]
[40]Tang J W,Ye J H 2005 Chem.Phys.Lett.410 104
[41]Zhao Z Y,Liu Q J 2008 J.Phys.D 41 025105
[42]Xun L,Dai L,Ma X G,Tang C Q,Tang D H 2007 Acta Phys. Sin.56 1048(in Chinese)[徐凌、戴磊、马新国、唐超群、唐代海2007物理学报56 1048]
[43]Peng L P,Xu L,Yin J W 2007 Acta Phys.Sin.56 1585(in Chinese)[彭丽萍、徐凌、尹建武2007物理学报56 1585]
[44]Jellison G E,Boatner L A,Budai J D,Budai J D,Jeong B S,Norton D P 2003 J.Appl.Phys.93 9537
[45]Zhu J F,Chen F,Zhang J l,Chen H J,Anpo M 2006 J. Photochem.Photobiol.A 180 196
[46]Ghasemi S,Rahimnejad S,Setayesh S R,Rohani S,Gholami M R 2009 J.Hazard Mater.172 1573
[47]Linsebigler A L,Lu G Q,Yates J T 1995 Chem.Rev.95 735
[48]Kim Y I,Atherton S J,Brigham E S,Mallouk T E 1993 Phys. Chem.97 11802
PACC:7115M,7115H,7115A
*Project supported by the National Natural Science Foundation of China(Grant No.50862009).
†E-mail:zxj4624@163.com
First-principles study on electronic structure and optical properties of anatase TiO2codoped with nitrogen and iron*
Zhang Xue-Jun1)2)†Gao Pan1)Liu Qing-Ju1)
1)(Key Laboratory of Nanomaterials&Nanotechnology of Yunnan Province,College of Physical Science and Technology,Yunnan University,Kunming650091,China)
2)(Department of Physics and Electric Information Engineering,Hunan City University,Yiyang413000,China)
(Received 13 October 2009;revised manuscript received 15 November 2009)
The crystal structure,electronic structure and optical properties of nitrogen and iron codoped anatase TiO2were studied by using the plane-wave ultrasoft pesudopotentials method based on density functional theory.The calculated results show that the octahedral dipole moments in nitrogen and iron codoped TiO2increase due to the changes in lattice parameters,bond length and charge of atoms,which is very effective for the separation of photoexcited electron-hole pairs and the improvement of the photocatalytic activity of TiO2.Some impurity energy levels of codoped TiO2are below the conduction band minimum,and others are above the valence band maximum.The distance between them is narrowed,which results in the redshift of the optical absorption edges to visible-light region.These impurity energy levels can reduce the recombination rate of photoexcited carriers and improve the photocatalytic efficiency of TiO2.Compared with that of Fe doped TiO2,for the codoped TiO2,the density of states peak of impurity energy levels above the valence band maximum increase apparently,which increases the electronic transition probability from the impurity energy levels to the conduction band,and improves the solar energy utilization.If the impurity level is not taken into account,compared with that of pure TiO2,the CB edge position and the VB edge position of codoped TiO2is only slightly changed,it means that the strong redox capacity of codoping photocatalysts is still excellent.
first-principles calculation,anatase TiO2codoped with nitrogen and iron,electronic structure,optical properties
book=344,ebook=344
*国家自然科学基金(批准号:50862009)资助的课题.
†E-mail:zxj4624@163.com
