LiFePO4中对位缺陷的第一性原理研究*
2010-09-08张华唐元昊周薇薇李沛娟施思齐
张华 唐元昊 周薇薇 李沛娟 施思齐
(浙江理工大学物理系,光电材料与器件中心,杭州310018)
(2009年12月12日收到;2009年12月26日收到修改稿)
LiFePO4中对位缺陷的第一性原理研究*
张华 唐元昊 周薇薇 李沛娟 施思齐†
(浙江理工大学物理系,光电材料与器件中心,杭州310018)
(2009年12月12日收到;2009年12月26日收到修改稿)
基于考虑了Fe-3d电子间的库仑作用U和交换作用J的GGA+U方案,应用第一性原理计算方法系统研究了LiFePO4的对位缺陷,以及对位缺陷的形成对材料的电导率和离子扩散速率的影响.结果表明,Li/Fe交换缺陷是最容易形成的,形成缺陷后的Fe—O键变长,扩宽了Li离子传输通道,有利于Li离子在通道中的扩散,对材料电化学性能的改善起到了一定的作用.
LiFePO4,对位缺陷,第一性原理计算
PACC:8630D,6170,3120G
1. 引言
锂离子电池与传统的二次电池相比保持了电压高、能量密度大、重量轻,又具有安全性能好、自放电小等优点[1],在实际生产生活中有着广泛的应用.橄榄石型结构的LiMXO4(M=Fe,Mn,Co,Ni;X =P,As,Si等)[2]是一系列非常有前景的锂离子电池正极材料.在LiMPO4这个家族里,LiFePO4的电化学性能是最好的[3],其容量高达170 mAh·g-1,放电平台是3.5 V,使用安全且价格低廉[4],具有优良的循环性能,可安全快速充放电,具有较高的振实密度,其质量、体积能量密度较高,热稳定性好,因此LiFePO4被认为是一种环境友好型正极材料,具有很好的发展前景,有可能在动力电池中得到广泛的应用,成为目前锂离子电池正极材料研究方向的热点[5].
1997年Goodenough等[6]开始研究并开发了磷酸盐材料作为锂离子电池的正极材料,其中LiFePO4成为锂离子电池正极材料研究的热点.但LiFePO4的电子电导率只有10-10—10-9S/cm,较差的电子迁移率和锂离子扩散速率成为限制其发展的主要因素,所以人们不断尝试去改善材料的电化学性能.从研究对材料进行碳包覆[7—11],到改变材料的颗粒大小[12],但碳包覆和改变颗粒的大小只是在一定程度上改善了LiFePO4的电导率,并不能从根本上改善材料的电化学性质.很多研究又对材料进行了锂位掺杂[13—15]和铁位掺杂[16—20]改性,并在实验中取得了很好的效果.目前,针对LiFePO4材料电子电导率的研究,其方法仍然是通过材料掺杂其他金属阳离子,期望能更进一步改善材料的电化学性能.
近年来,利用第一性原理计算对LiFePO4材料的磁性、结构、表面和晶格动力学进行研究都有了一定的进展[21—24].材料的缺陷问题是一个比较有意义的课题,人们还没有完全对其认识清楚.本文基于考虑了Fe-3d电子间的库仑作用U和交换作用J的GGA+U方案,应用密度泛函理论(DFT)的第一性原理计算方法系统研究LiFePO4的对位缺陷.主要讨论的是Fe占据Li位的对位缺陷,以及缺陷的形成对材料电导率和离子迁移率产生的影响,并和相关文献的实验结果进行了比较.基于这一比较系统的理论计算工作,可望对实验工作有较好的指导作用.
2. 计算方法
LiFePO4在自然界中以磷铁锂矿的形式存在(见图1),为橄榄石型结构,属于正交晶系,空间群为Pnma.实验测定的晶格常数a=10.3290(1=0.1nm),b=6.0065,c=4.6908[25].其P原子占据四面体的4c位,Fe原子和Li原子分别占据八面体的4c和4a位.以b轴方向为视角,可以看到FeO6八面体在bc平面上以一定的角度连接起来,而LiO6八面体则沿b轴方向相互共边,形成链状.每个FeO6八面体分别与一个PO4四面体和两个LiO6八面体共边.同时,每个PO4四面体还与两个LiO6八面体共边.正是LiO6八面体沿着b轴方向形成链状导致了沿着b方向存在可容许锂离子穿过的通道.图1(b)表示在橄榄石结构中,与[001]hexagonal垂直的密堆积氧层之间只存在一种类型的阳离子排列方式.
计算应用的是基于密度泛函理论的第一性原理方法,离子核和电子之间的相互作用采用凝聚芯投影缀加波法(PAW)[26].使用的计算软件包是VASP[27—31].Li-2s12p0,O-2s22p4,P-3s23p3,Fe-3d74s1被视为价电子构型,平面波的能量截断为520 eV,采用1×1×1 Monkhorst-Pack类型的k点进行布里渊区的积分,其高斯展宽为0.20 eV[32],这组参数保证了总能量收敛于每个原子能量为2 meV的水平上[33].在优化结构中,超原胞中的原子是弛豫的,晶格参数固定在化学单胞的理论计算值,当作用于所有弛豫原子的力小于0.02 eV/时,计算停止.采用广义梯度近似(GGA)[28]描述电子交换-关联能.为描述Fe-3d电子之间的库仑作用,用Liechtenstein等[29]提出的旋转不变方法(Rotationally Invariant Approach)在GGA中增加了Hubbard参数U(GGA+U),这里库仑作用U和交换作用J是独立的参数.U和J的值分别取为4.7和1.0 eV,它是由Zhou等[30]用Cococcioni和de Gironcoli[31]提出的线性回归方法通过自洽计算而得到的.

图1 LiFePO4结构示意图(黑点为O原子,灰点为Li原子)(a)单胞(AOB代表直角坐标系),(b)PO4四面体,(c)FeO6六面体
3. 结果与讨论
3.1. 三种对位缺陷的形成能
我们所定义的对位缺陷,主要考虑三种情况.第一种是替换缺陷,即体系外的Fe替换LiFePO4中的Li;第二种是Li空位缺陷,即体系内Fe占据Li位,在原Fe位形成空位;第三种是锂铁交换缺陷,即体系内部Li,Fe进行位置交换.三种对位缺陷类型的定义和形成能公式分别如下.
3.1.1. 替换缺陷
1)不考虑价态平衡的条件下(替换缺陷-1):

2)考虑价态平衡的条件下(替换缺陷-2):


3.1.2. 锂空位缺陷
1)不考虑价态平衡的条件下(空位缺陷-1):
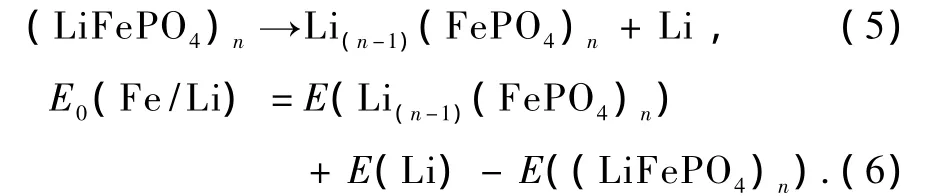
2)考虑价态平衡的条件下(空位缺陷-2):

3.1.3. 锂铁交换缺陷(一种特殊的对位缺陷,也即二次对位缺陷)

锂铁交换缺陷由于位置不同则考虑两种情况,交换缺陷-1是Fe占据Li位(1/2,1/2,1/2),交换缺陷-2是Fe占据Li位(1/2,1/4,1/2).
我们基于2×2×2的超晶胞(8个LiFePO4,224个原子)构建对位缺陷,计算缺陷的形成能.根据实验数据得知,内部缺陷的浓度必须小于0.3%[34],因此我们选择只替换一个原子,即构建一个内部缺陷.在优化了结构并计算得到对位缺陷各种类型形成前后体系的总能量后,我们能给出各种缺陷的体系原子数和形成能以及由此导致的键长变化,分别如表1和表2所示.根据缺陷形成能数据进行比较,在LiFePO4超晶胞体系中,替换缺陷和锂空位缺陷这两种对位缺陷都很难形成,尤其是空位缺陷.而交换缺陷类型中Fe占据Li(1/2,1/2,1/2)位置(交换缺陷-1)的缺陷形成能是最低的(0.845 eV),这与可获得的实验值(0.74 eV)[35]符合得非常好.可以说明,交换缺陷是最容易形成的对位缺陷.

表1 化学单胞LiFePO4及包含各种缺陷的超原胞中的原子数和缺陷形成能计算值

表2 超晶胞LiFePO4各种对位缺陷形成前后Li离子通道中的原子键长数据
表2所列举的是超晶胞LiFePO4体系中心Li离子通道中缺陷前(Li原子)和缺陷后(Fe原子)测得的键长数据,从表2中的数据得知,形成缺陷以前,中心Li离子通道中Li—O键的键长是2.149,当交换缺陷形成后Fe占据Li位,其Fe—O键的键长是2.187,与其他对位缺陷相比较键长变化是最大的.从直观角度来说(见图2),交换缺陷形成后,中心Li离子通道中的Fe—O键的键长变长,则Li离子通道通过缺陷就会变宽,文献[36—38]验证了Li离子的迁移是沿着LiFePO4中的[010]通道的,并不能在两个Li离子通道之间跳跃,所以可以推断这必将有利于Li离子的嵌入和脱出,进而提高了Li离子在LiFePO4中的迁移率.
3.2. 缺陷形成后的电子结构
过渡金属氧化物的性能与电子结构有着密切的关系,为了理解内部缺陷形成后对LiFePO4材料的电子结构产生的影响,我们对纯的LiFePO4和对位缺陷类型中最易形成的交换缺陷的电子结构进行了深入的分析.
电子态密度能反映材料的电子结构情况.首先,我们来分析一下纯的LiFePO4的电子结构.图3是纯的LiFePO4的分波态密度(PDOS)图,从图3中可以看到,Fe-3d中自旋向上与自旋向下的部分在费米能级附近产生了明显的分裂.分裂来自于电子的排布需满足第一条洪特规则的限制.自旋向上的部分全部被填充.对于自旋向下的部分,在LiFePO4中,仅仅在费米能级以下的0.28 eV处有一个峰.其他的都没有被填充.由于Fe离子是处在由O离子构成的近似八面体中心,因此本来五重简并的Fe-3d态在该八面体晶体场的作用下分裂成三重简并的t2g态(dxy,dyz和dxz)和二重简并的eg(dx2-y2和dz2)态,而且对称度的降低和其与O-2p态的轨道作用会导致3d态的进一步分裂.

图3 体相LiFePO4中Fe(实线)和O(虚线)的分波电子态密度零点为费米能级
费米能级附近的窄带是Fe的3d部分,Fe的非成键t2g态和反成键eg态分别在1.8—3.1 eV和 3.4—4.0 eV的能量范围,这种占据方式表明其中的Fe离子呈典型的+2价,与其结构价态一致.
图4分别给出了纯的和包含Li/Fe交换缺陷的LiFePO4的总电子态密度(TDOS)图,可以清晰地看出,纯的LiFePO4和交换缺陷形成后的LiFePO4均为半导体,电子态密度的整体峰型基本上是一致的,费米能级仍位于带隙的中央,电子在各能级的占据情况基本相同.从数值上看,纯的LiFePO4的带隙约为3.1 eV,和文献报道的实验数据3.7 eV[35]较为接近.对位缺陷的带隙是2.9 eV,和纯的LiFePO4相比,带隙略微变窄,但是其变化不太明显,所以对材料的电子电导率不会产生太大的影响.但至少可以说明,交换缺陷形成后,对材料的电子电导并没有起到阻碍的作用.

图4 纯的LiFePO4(虚线)和Li/Fe交换缺陷的LiFePO4(实线)总电子态密度图零点为费米能级
4. 结论
本文采用基于密度泛函理论的第一性原理计算方法,系统地研究了LiFePO4内部缺陷的形成情况,其结果表明:
1)LiFePO4对位缺陷中,交换缺陷(交换缺陷-1)的形成能是最低的.
2)形成交换缺陷后键长的改变是最大的,因此我们可以预测,由于Fe—O键长的变大,扩宽了LiFePO4中[010]方向上的一维Li离子传输的通道,我们认为,这样有利于Li离子的嵌入和脱出,有利于LiFePO4中Li离子电导率的提高.
3)LiFePO4中的交换缺陷,对其电子电导并没有起到阻碍的作用.
[1]Hou Z F,Liu H Y,Zhu Z Z,Huang M C,Yang Y 2003 Acta Phys.Sin.52 952(in Chinese)[侯柱峰、刘慧英、朱梓忠、黄美纯、杨勇2003物理学报52 952]
[2]Amin R,Lin C,Maier J 2008 Phys.Chem.Chem.Phys.10 3524
[3]Huang H,Yin S C,Nazar L F 2001 Electrochem.Solid-State Lett.4 A170
[4]Islam M S,Driscoll D J,Fisher C A J,Slater P R 2005 Chem. Mater.17 5085
[5]Sigle W,Amin R,Weichert K,Aken P A,Maier J 2009 Electrochem.Solid-State Lett.128 A151
[6]Padhi A K,Nanjundaswamy K S,Masquelier C,Okada S,Goodenough J B 1997 J.Electrochem.Soc.144 1188
[7]Ravet N,Chouinard Y,Magnan J F,Besner S,Gacuthier M,Armand M 2001 J.Power Sources 503 97
[8]Dominko D,Gaberscek M,Drofenik J,Bele M,Jamnik J 2003 Electron.Acta 48 3709
[9]Choi D,Kumta P 2007 J.Power Sources 163 1064
[10]Chung S Y,Bloking J T,Chiang Y M 2002 Nat.Mater.1 123
[11]Chung S Y,Chiang Y M 2003 Electrochem.Solid-State Lett.6 A278
[12]Herle P S,Ellis B,Coombs N,Nazar L F 2004 Nat.Mater.3 147
[13]Barker J,Saidi M Y,Swoyer J L 2003 Electrochem.Solid-State Lett.6 A53
[14]Islam M S,Driscoll D J,J Fisher C A,Slater P R 2005 Chem. Mater.17 5085
[15]Shi S Q,Ouyang C Y,Wang D S,Wang Z X,Chen L Q 2003 Phys.Rev.B 68 19508
[16]Hong J,Wang C,Kasavajjula U 2006 J.Power Sources 162 1289
[17]Mi C H,Zhang X G,Zhao X B,Li H L 2006 Mater.Sci.Eng. B 8 129
[18]Wang D Y,Li H,Shi S Q,Huang X J,Chen L Q 2005 Electron.Acta 50 2955
[19]Wang G X,Needham S,Yao J,Wang J Z,Liu R S,Liu H K 2006 J.Power Sources 159 282
[20]Ouyang X F,Shi S Q,Ouyang C Y,Jiang D Y,Liu D S,Ye Z Q,Lei M S 2007 Chin.Phys.Soc.10 3042
[21]Shi S Q,Ouyang C Y,Xiong Z H,Liu L J,Wang Z X,Li H,Wang D S,Chen L Q,Huang X J 2005 Phys.Rev.B 71 144404
[22]Luo Z,Di N L,Kou Z Q,Cheng Z H,Liu L J,Chen L Q,Huang X J 2004 Chin.Phys.13 2158
[23]Shi S Q,Zhang H,Ke X Z,Ouyang C Y,Lei M S,Chen L Q 2009 Phys.Lett.A 44 4906
[24]Ouyang X F,Lei M L,Shi S Q,Luo C L,Liu D S,Jiang D Y,Ye Z Q,Lei M S 2009 J.Alloys Compd.476 462
[25]Liu H,Xie J Y 2009 J.Mater.Process.Technol.209 477
[27]Kresse G,Furthmüller J 1996 Phys.Rev.B 54 11169
[28]Perdew J P,Chevary J A,Vosko S H,Jackson K A,Pederson M R,Singh D J,Fiolhais C 1992 Phys.Rev.B 46 6671
[29]Liechtenstein A I,Anisimov V I,Zaanen J 1995 Phys.Rev.B 52 R5467
[30]Zhou F,Kang K,Maxisch T,Ceder G,Morgan D 2004 Solid State Commun.132 181
[31]Cococcioni M,de Gironcoli S 2005 Phys.Rev.B 71 035105
[32]Methfessel M,Paxton A T 1989 Phys.Rev.B 40 3616
[33]Huang Y X,Cao Q X,Li Z M,Li G F,Wang Y P,Wei Y G 2009 Acta Phys.Sin.58 8002(in Chinese)[黄云霞、曹全喜、李智敏、李桂芳、王毓鹏、卫云鸽2009物理学报58 8002]
[34]Fisher C A J,Hart Prieto V M,Saiful Islam M 2008 Chem. Mater.20 5907
[35]Islam M S 2005 Chem.Mater.17 5085
[36]Ouyang C Y,Shi S Q,Wang Z X,Huang X J,Chen L Q 2004 Phys.Rev.B 69 104303
[37]Ouyang C Y,Shi S Q,Wang Z X,Li H,Huang X J,Chen L Q 2004 J.Phys.:Condens.Matter 16 2265
[38]Morgan D,van der Ven A,Ceder G 2004 Electrochem.Solid-State Lett.7 A30
PACC:8630D,6170,3120G
*Project supported by the National Natural Science Foundation of China(Grant No.50802089),the“Qianjiang Talent Project”of Zhejiang Province,China(Grant No.2007R10028),the Scientific Research Starting Foundation for the Returned Overseas Chinese Scholars of Ministry of Education of China(Grant No.[2008]890),the Natural Science Foundation of Zhejiang Province,China(Grant No.Y4090280),and the Natural Science Foundation of Jiangxi Province,China(Grant No.2008GZW0014).
†Corresponding author.E-mail:siqishihz@gmail.com
Antisite defect of LiFePO4:A first-principles study*
Zhang Hua Tang Yuan-Hao Zhou Wei-Wei Li Pei-Juan Shi Si-Qi†
(Department of Physics,Center for Optoelectronics Materials and Devices,Zhejiang Sci-Tech University,Hangzhou310018,China)
(Received 12 December 2009;revised manuscript received 26 December 2009)
The antisite defect,electronic conductivity and ionic dynamic properties of LiFePO4have been investigated using first-principles density functional theory taking into account the on-site Coulomb interaction within the GGA+U scheme. Results indicate the Li/Fe exchange defect is the most preferred to occur in LiFePO4,which causes the Fe—O bond length to change in the direction favoring the formation of Li+diffusion channels,hence improving the ionic dynamic properties of the olivine LiFePO4.
LiFePO4,antisite defect,first-principles calculations
book=282,ebook=282
*国家自然科学基金(批准号:50802089)、浙江省“钱江人才计划”(批准号:2007R10028)、教育部留学回国人员科研启动基金(批准号:[2008]890)、浙江省自然科学基金(批准号:Y4090280)和江西省自然科学基金(批准号:2008GZW0014)资助的课题.
†通讯联系人.E-mail:siqishihz@gmail.com
