Zr 催化剂对NaAlH4和Na3AlH6可逆储氢性能的影响*
2010-09-08叶佳宇刘亚丽王靖林何垚
叶佳宇 刘亚丽 王靖林 何垚
(云南大学物理系,昆明650091)
(2009年6月28日收到;2009年11月20日收到修改稿)
Zr 催化剂对NaAlH4和Na3AlH6可逆储氢性能的影响*
叶佳宇 刘亚丽 王靖林 何垚†
(云南大学物理系,昆明650091)
(2009年6月28日收到;2009年11月20日收到修改稿)
采用基于密度泛函理论的平面波赝势方法,分别计算纯净的以及掺杂Zr的NaAlH4和Na3AlH6的晶格结构常数、能量、电子局域函数和电子态密度.结果表明:NaAlH4和Na3AlH6分别是带隙为4.6和3.1 eV的绝缘体; NaAlH4和Na3AlH6中Al—H键是共价键,Na—H键是离子键;Zr原子替代Na原子后,NaAlH4中Zr—H键比原Na—H键强,同时Al—H键变弱;Zr原子替代Al原子后,Zr—H键比原Al—H键弱,使H原子容易脱离.从结合能看,NaAlH4和Na3AlH6掺杂Zr后,结构比原来稳定,脱氢需要的能量降低.
储氢,NaAlH4,Na3AlH6,Zr掺杂
PACC:7115M,7125
1. 引言
氢在21世纪作为一种重要的能源已经成为人们的共识,但却没有能在工业能源中得到广泛应用,根本的原因在于氢气存储的问题难以解决.因此,对性能优越、安全性高的储氢材料的研究已经成为物理学、化学、材料科学等学科共同关心的一个热点.目前正在研究的储氢材料主要有金属氢化物、配位氢化物、纳米材料、多孔吸附储氢材料等.例如:戴伟等[1]通过蒙特卡罗模拟得出孔结构是决定储氢吸附量大小的关键因素.易双萍等[2]实验发现碳纳米管的热处理温度对其电化学储氢性能有着较大的影响.陈玉红等[3]用密度泛函理论(DFT)的方法预测了[Mg(NH2)2]n(n=1—5)各团簇的最稳定结构,并对最稳定结构的振动特性、成键特性、电荷特性等进行了理论研究.周晶晶等[4]采用第一性原理研究了近几年涌现出的一批新型轻质储氢材料,得出如下结论:碳纳米管、金属有机框架多孔材料和C60通过增大化学吸附成键的键能可改善吸放氢量,Al/B系复杂氢化物和金属-氮-氢系通过降低结合能也可改善放氢温度.在各种储氢材料中,由[AlH4]-,[NH2]-和[BH4]-生成的盐作为储氢材料被称为“配位氢化物”(complex hydrides),它们都有很高的储氢质量密度,这些储氢量高、成本低的储氢材料是当前的一个重要研究方向[5].
近年来对于储氢材料NaAlH4的研究越来越多,NaAlH4被认为是最有希望的车载燃料电池材料之一[6],其含氢量高达7.4%.NaAlH4通过以下两步进行中低温下脱氢反应[7,8]:

第一步分解反应在185—230℃放出3.7%氢气,第二步在大约260℃放出1.9%氢气,理论放氢量达5.6%,达到国际能源署对储氢材料提出的目标.但纯净的NaAlH4分解所需要的高温条件和分解反应的不可逆性,一直制约着NaAlH4作为储氢材料的发展.
1997年,Bogdanovic等[9]研究发现,在NaAlH4中掺入摩尔分数为2%的Ti离子后,体系的放氢反应在160℃即可完成,动力学性能也得到改善,更重要的是实现了NaAlH4的可逆储氢.随后的研究发现添加Zr离子也有助于提高NaAlH4循环的动力学性能,并且Ti和Zr具有良好的兼容性,可一起应用于优化吸/脱氢反应.加入Ti-Zr合金氢化物的NaAlH4具有良好的储放氢动力学性能,在160℃,0.1 MPa放氢条件下,其总放氢量达4.5%,且具有良好的循环稳定性[10].Zidan等[11]发现,用Ti和Zr的醇盐共掺作为提高NaAlH4体系活性的催化剂,Zr离子对第一步分解反应的催化效果没有Ti离子好,但对于第二步降低反应的分解温度比Ti离子明显.研究发现,过渡金属能提高配位氢化物储氢性能. Wang等[12]通过实验计算了不同比例的Sc掺杂对NaAlH4循环储氢量的影响,并与掺杂Ti进行了对比,发现掺杂Sc比掺杂Ti的动力学性能和储氢量高,但所需温度也高很多.Naik等[13]也对几种不同的过渡金属(ScCl3,TiCl3,VCl3和MnCl2)对NaAlH4动力学性能的影响进行了实验研究.Marashdeh等[14]研究指出,Ti掺杂的最佳位置是替代晶体表面的Na原子.Araújo等[15]用第一性原理计算Ti掺杂NaAlH4前后能量和电荷分布的变化.Li等[16]分析了在Na3AlH6中Ti分别替代Na或Al原子,以及在Na或Al处产生空穴时能量的变化.有关Zr掺杂的理论研究作者至今尚未见文献报道.本文根据第一性原理优化NaAlH4和Na3AlH6的晶格结构,计算Zr原子替代Al原子或Na原子前后的能量、电子局域函数(ELF)、态密度的变化,并对计算结果进行了分析比较.
2. 计算方法
在DFT[17]框架下交换关联项,使用广义梯度近似(GGA)下由Perdew,Burke和Ernzerhof[18]提出的PBE泛函形式,外层电子与内层原子芯核的相互作用用投影缀加波[19],平面波能量截断取250 eV,k点选取3×3×3模式,原子结构优化中的总能收敛性判据为10-4eV,具体计算采用VASP软件包[20,21].计算中,采用2×2×1的96和80个原子的超晶胞模型,未掺杂及掺杂Zr的NaAlH4超晶胞模型分别用Na16Al16H64,(Na15Zr)Al16H64和Na16(ZrAl15)H64表示,未掺杂及掺杂Zr的Na3AlH6超晶胞模型分别用Na24Al8H48,(Na23Zr)Al8H48和Na24(ZrAl7)H48表示.
3. 计算结果与讨论
3.1. 结构优化
常温常压下,X射线衍射测得NaAlH4属于I41/a空间群,其中Al原子占据4b位,Na原子占据4a位,H原子占据16f位[22],其结构如图1(a)所示.首先对NaAlH4和Na3AlH6晶体结构参数进行优化,为了便于比较,表1列出了本文的计算结果和文献[23]的计算结果,同时也一并列出了文献[22,24]的实验值.优化后晶格常数a=0.500nm,c= 1.108nm,与实验值[22]a=0.498nm,c=1.115nm非常接近,仅相差约0.56%.Na3AlH6属于P21/n空间群,其中Al原子占据2a位,Na原子占据2b和4e位[24],6个H原子在Al原子周围形成八面体的[AlH6]3-,其结构如图1(b)所示.优化后晶格常数a=0.533nm,b=0.554nm,c=0.770nm,与实验值[24]a=0.541nm,b=0.554nm,c=0.777nm相差约0.83%.
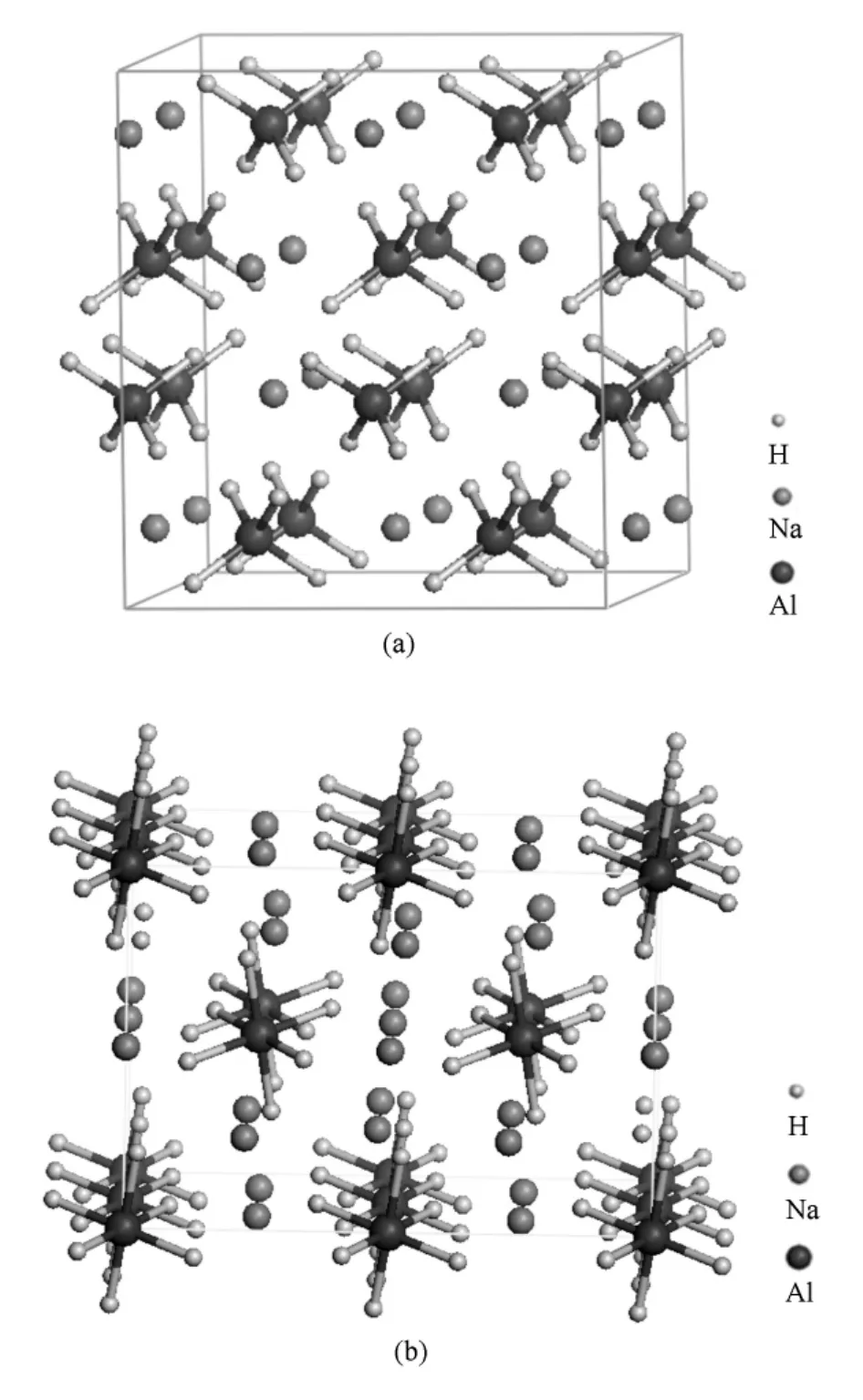
图1 NaAlH4和Na3AlH6的晶体模型(a)NaAlH4,(b)Na3AlH6

表1 NaAlH4和Na3AlH6的晶格常数及体积
为了判断Zr原子替代Na原子或Al原子后结构稳定性的变化,找出Zr掺杂的最佳位置,我们通过下式[16]计算掺杂前后的形成焓:
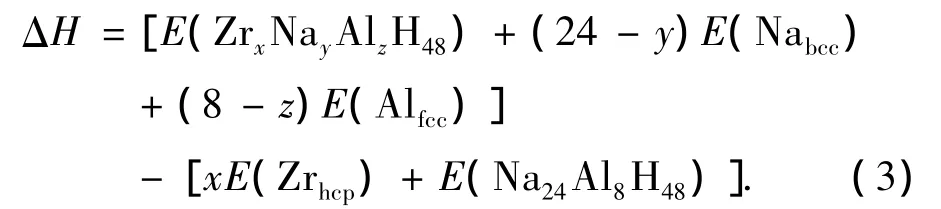
这里x,y,z分别代表Zr,Na,Al原子数.我们用Zr,Na,Al原子的结合能作为参数计算ΔH,得到六角密堆积结构的Zr原子、体心立方结构的Na原子和面心立方结构的Al原子的能量分别是6.448,1.091和3.475 eV,相应的实验值为[25]6.25,1.113和3.39 eV,H2分子的结合能为4.482 eV,实验值为[26]4.75 eV,计算结果与实验结果符合很好.按照(3)式计算得出的能量列于表2,分别取NaAlH4和Na3AlH6的形成焓为零作为参照,计算得到的ΔH为负值,表示Zr原子替代Na原子或Al原子比纯净的NaAlH4和Na3AlH6更稳定.从表2可以看出,掺杂Zr比纯净的NaAlH4和Na3AlH6更稳定,而Zr替代Na比替代Al的结构稳定性更好.
从未掺杂和掺杂Zr的晶胞中移去一个H原子所需要的能量为[17]
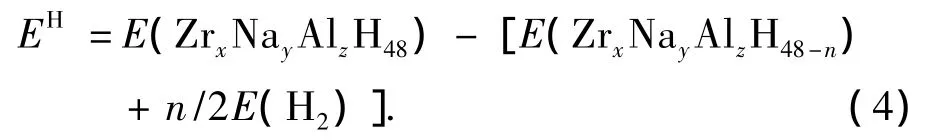
在计算中重新优化Na16Al16H63,(Na15Zr)Al16H63,Na16(ZrAl15)H63,Na24Al8H47,(ZrNa23)Al8H47和Na24(ZrAl7)H47的结构并分别计算能量,所得结果列于表2.从表2不难看出,NaAlH4移去一个H原子需要4.767 eV,比Na3AlH6移去一个H原子所需能量大0.678 eV,说明NaAlH4中Al—H键比Na3AlH6中Al—H键强.Zr替代Na原子或Al原子后,NaAlH4和Na3AlH6脱氢所需的能量减小,因此,掺杂Zr使NaAlH4分解放氢的温度降低.

表2 不同超晶胞结构的形成焓ΔH和脱一个H原子需要的能量EH
3.2. ELF
为了分析原子之间相互作用的本质,我们对各原子之间的电子局域性进行了计算.图2给出了未掺杂的NaAlH4以及Zr原子替代Na原子和Al原子的NaAlH4在(001)面同一位置处的ELF图,灰度条的左端(0)代表电子局域性最弱,灰度条的右端(1)代表电子局域性最强,0.5对应于均匀电子气,由黑到白电子局域性从0渐变到1.通过分析ELF图可以了解原子之间成键的本质.从图2(a)不难看出,AlH4络合物内部Al原子与H原子之间的电子局域性很强,因此它们之间形成共价键,而Na原子与近邻的H原子之间的电子局域性很弱,形成离子键.当用Zr原子替代Na原子之后,各原子之间的电子局域性发生了显著的改变(图2(b)),Zr与周围[AlH4]-空隙中ELF值升高(0.3—0.6之间),这是典型的金属键.Zr—AlH4键之间的变化是由于Zr的电离能(640.1 kJ/mol)比Na的电离能(495.8 kJ/mol)高,所以Na比Zr容易失去电子,同时Zr原子附近AlH4络合物内部的电子局域性比图2(a)中降低,说明Al原子与H原子之间的共价键变弱.图2(c)中电子局域性变化没有图2(b)中那样明显,但还是可以看出Zr原子与H原子之间电子局域性比纯净的NaAlH4中Al原子与H原子之间的电子局域性降低.同时我们还计算了键长的变化,NaAlH4中Al—H键键长为0.164nm,Na—H键键长为0.240nm,用Zr原子替代Na原子后,Zr原子周围AlH4络合物中Al—H键键长变为0.172nm,Zr—H键键长为0.211nm,说明Zr原子吸引近邻的H原子,使Al—H键变长,减弱了Al—H键的强度;而Zr原子替代Al原子后的NaAlH4中Zr—H键键长为0.198nm,邻近Na—H键键长为0.216nm,Zr—H键比替代前的Al—H键长.因此,当Zr原子替代Na原子后,Al—H键的强度减弱,Zr原子替代Al原子后,Zr—H键比原Al—H键弱,使得H原子更容易从AlH4络合物中分解出来,于是NaAlH4的放氢反应完成所需的温度会降低,动力学性能也得到了改善.

图2 未掺杂和掺杂后的NaAlH4在(001)面的ELF图(a)纯净的NaAlH4,(b)Zr原子替代Na原子的NaAlH4,(c)Zr原子替代Al原子的NaAlH4
图3给出了纯净的Na3AlH6以及Zr原子替代Na原子和Al原子的Na3AlH6在(001)面同一位置处的ELF图.从图3(a)可以看出,Na3AlH6与NaAlH4相同,AlH6络合物内部Al原子与H原子之间形成共价键,而Na原子与H原子之间是离子键.图3(b)中,Zr原子替代Na原子,Zr原子吸引周围6个AlH6络合物中邻近的H原子,使邻近的Al—H键键长由0.176nm增加到0.183nm.Zr与邻近H原子形成八面体结构,Zr—H键键长为0.209nm,小于原Na3AlH6中Na—H键的键长(0.222nm).Zr原子替代Al原子后,晶胞体积比纯净Na3AlH6更大,这是由于Zr—H键的键长为0.200nm,远远大于原Al—H键的键长(0.176nm).应当注意的是,只有Zr原子替代Al原子的离子团键长发生了变化,其他AlH6络合物结构几乎没有变化.从图3也可以看出,Zr掺杂Na3AlH6的ELF图变化没有NaAlH4那么明显,但是从键长的变化仍然可以得出,Zr原子替代Na原子和Al原子后,Al—H键的强度减弱,使得H原子更容易从AlH6络合物中分解出来.

图3 未掺杂和掺杂后的Na3AlH6在(001)面的ELF图(a)纯净的Na3AlH6,(b)Zr原子替代Na原子的Na3AlH6,(c)Zr原子替代Al原子的Na3AlH6
3.3. 电子态密度
图4、图5和图6分别给出纯净的NaAlH4、Zr原子替代Na原子和Al原子的NaAlH4超晶胞的总态密度(TDOS)和分波态密度(PDOS),定义费米能级EF=0 eV.图5给出了(Na15Zr)Al16H64的TDOS以及距离Zr原子最近的Al原子和H原子的PDOS,图6给出了Na16(ZrAl15)H64的TDOS以及距离Zr原子最近的Na原子和H原子的PDOS,图4与Araújo等[15]得到的态密度图相同.从图4不难看出NaAlH4是绝缘体,导带与价带之间的禁带宽度为4.6 eV.NaAlH4的价带主要分布范围为-6—0 eV,导带分布在4.6—8 eV之间.价带顶部态密度主要是由H的1s态和Al的3p态轨道杂化形成,费米能级以上导带底部的态密度主要由Na和Al的s和p态组成(图4).Zr替代Na后,TDOS发生了明显变化.随着Zr的引入,在带隙中产生两个峰并引起带隙减小(图5),这两个峰主要是Zr的d轨道与Al和H的电子轨道杂化而成,与上述ELF图(图2 (b))Zr与AlH4之间不是离子键的结论相符.从图5还可以看出,掺杂Zr使Al和H的电子轨道杂化程度降低,Al—H键减弱,但掺杂后NaAlH4态密度的价带部分仍然是由H的1s态和Al的3s3p态组成,因此Al—H键仍然是共价键.Zr原子替代Al原子后,在带隙中引入两个峰并使带隙减小,Zr与H的电子轨道杂化程度比纯净NaAlH4中Al与H的电子轨道杂化程度明显减少(图6),因此Zr—H键比Al—H键减弱很多.

图4 Na16Al16H64的TDOS和PDOS

图5 (Na15Zr)Al16H64的TDOS和PDOS
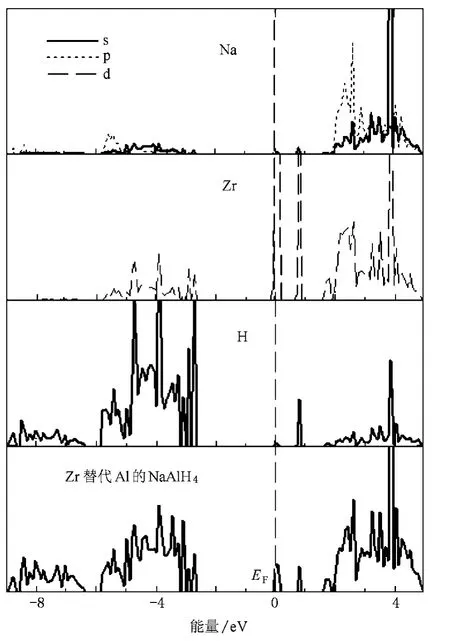
图6 Na16(ZrAl15)H64的TDOS和PDOS

图7 Na24Al8H48的TDOS和PDOS
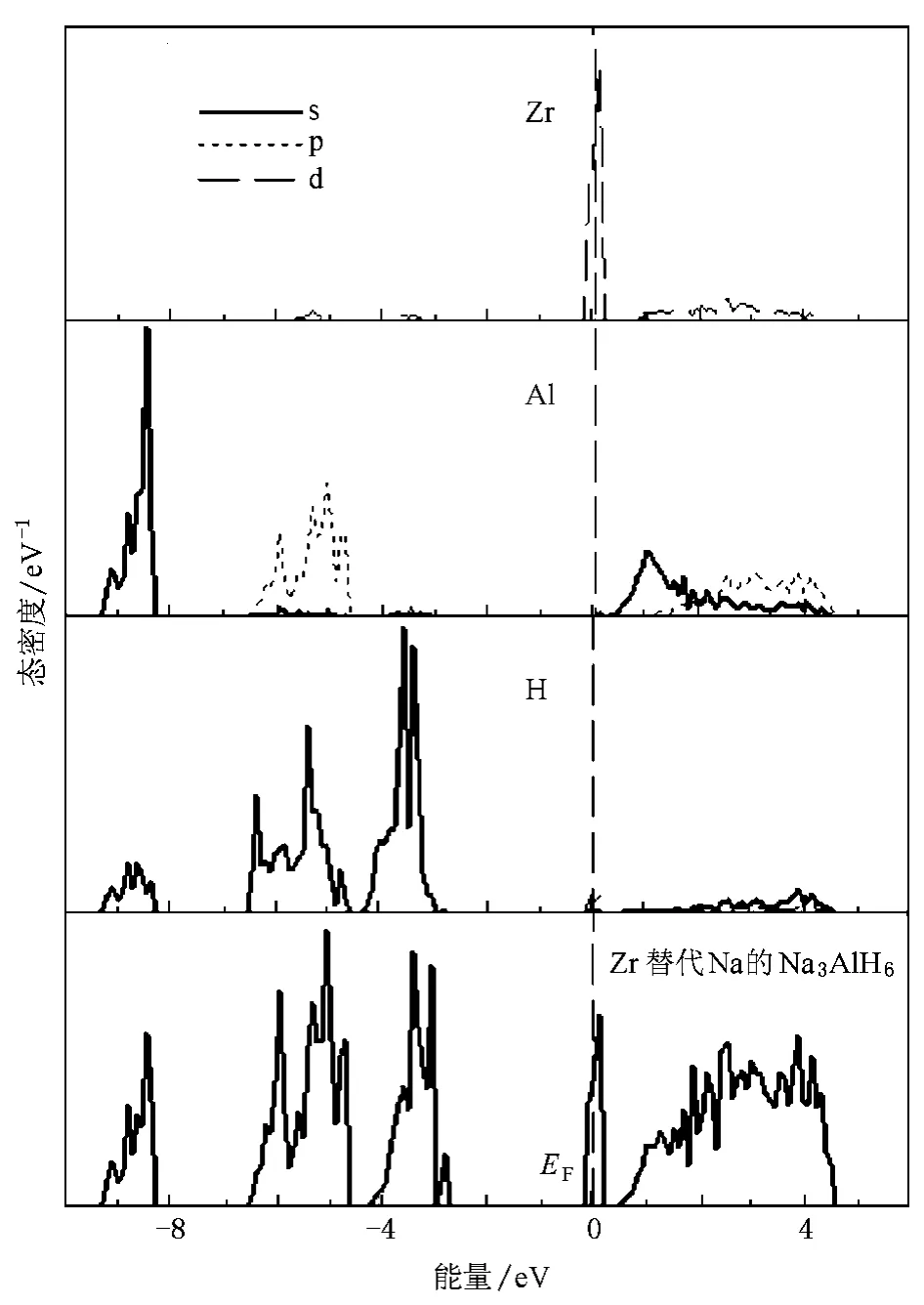
图8 (Na23Zr)Al8H48的TDOS和PDOS

图9 Na24(ZrAl7)H48的TDOS和PDOS
图7、图8和图9分别给出了未掺杂的Na3AlH6、掺杂Zr的Na3AlH6超晶胞的TDOS和PDOS,定义费米能级EF=0 eV.图8给出了(Na23Zr)Al8H48的TDOS以及距离Zr原子最近的Al和H原子的PDOS,图9给出了Na24(ZrAl7)H48的TDOS以及距离Zr原子最近的Na和H原子的PDOS,图7与文献[16]得到的态密度图相同.从图7可得出Na3AlH6是带隙为3.1 eV的绝缘体,比NaAlH4的带隙窄1.5 eV.与NaAlH4不同,Na3AlH6的价带分为三部分.最下面的价带由Al和H的s轨道杂化形成,能量分布在-6.5—-5.6 eV之间,间隔大约为2 eV,是由H的1s态和Al的3p态组成的中间价带,最上面的价带由H的s态单独形成.费米能级以上导带底部的态密度主要是由Na的s和p轨道形成的反键态(图7).当用Zr原子替代Na原子后,Zr的d轨道在导带底部的费米能级处产生了一个尖峰,但Zr原子的d轨道与H原子的s轨道没有相互作用,它们之间不成键态,并且Zr的引入对Al和H态密度分布没有产生太大的影响(图8).当用Zr原子替代Al原子后,Zr原子的d轨道分裂成了eg和t2g轨道,于是Zr原子的eg轨道可以与H原子的s轨道杂化形成成键态,TDOS在-4 eV附近产生了一个峰,并且Zr原子的s,p,d轨道在价带和导带都有明显的态分布,都与H的s轨道杂化.同时Zr原子周围H原子的态密度发生很大变化,H原子的PDOS主要分布在价带中部-3—-6 eV之间(图9),而不是分成三个价带.从上述的Na3AlH6的ELF图中(图3(b))也不难看出,Zr原子替代Na原子后虽然吸引周围AlH6络合物中的H原子,但Zr原子与H原子之间并没有形成共价键,而Zr原子替代Al原子后与周围H原子之间形成的是共价键.
4. 结论
本文采用第一性原理计算系统地研究了Zr原子掺杂到NaAlH4和Na3AlH6两种体系后的能量、ELF和电子态密度的变化,分析了掺杂Zr原子对提高NaAlH4动力学性能和降低分解温度的原因.Zr原子替代NaAlH4和Na3AlH6中的Na原子后,会减弱原来的Al—H键的强度,使H原子更容易从AlH4和AlH6络合物中脱离;用Zr原子替代Al原子后形成的Zr—H键键长远大于原Al—H键键长,Zr—H键的强度比Al—H键弱.掺杂Zr使晶胞结构更稳定,脱氢所需能量也降低.因此,掺杂Zr提高了NaAlH4循环储氢的动力学性能.
[1]Dai W,Luo J S,Tang Y J,Wang C Y,Chen S J,Sun W G 2009 Acta Phys.Sin.58 1890(in Chinese)[戴伟、罗江山、唐永建、王朝阳、陈善俊、孙卫国2009物理学报58 1890]
[2]Yi S P,Zhang H Y,Ouyang Y,Wang Y H,Pang J S 2006 Acta Phys.Sin.55 2644(in Chinese)[易双萍、张海燕、欧阳玉、王银海、庞晋山2006物理学报55 2644]
[3]Chen Y H,Kang L,Zhang C R,Luo Y C,Ma J 2008 Acta Phys.Sin.57 4866(in Chinese)[陈玉红、康龙、张材荣、罗永春、马军2008物理学报57 4866]
[4]Zhou J J,Chen Y G,Wu C L,Zheng X,Fang Y C,Gao T 2009 Acta Phys.Sin.58 4853(in Chinese)[周晶晶、陈云贵、吴朝玲、郑欣、房玉超、高涛2009物理学报58 4853]
[5]Orimo S I,Nakamori Y,Eliseo J R,Zuttel A,Jense C M 2007 Chem.Rev.107 4111
[6]Schuth F,Bogdanovic B,Felderhoff M 2004 Chem.Commun. 20 2249
[7]Ashby E C,Kobetz P 1966 Inorg.Chem.5 1615
[8]Dilts D A,Ashby E C 1972 Inorg.Chem.11 1230
[9]Bogdanovic B,Schwickardi M 1997 J.Alloys Compd.253 1
[10]Zhuang P H,Liu X P,Li Z N,Wang S M,Jiang L J,Li H L 2008 Chin.J.Nonferr.Met.18 671(in Chinese)[庄鹏辉、刘晓鹏、李志念、王树茂、蒋利军、李华玲2008中国有色金属学报18 671]
[11]Zidan R A,Takara S,Hee A G,Jensen C M 1999 J.Alloys Compd.285 119
[12]Wang T,Wang J,Ebner A D,Ritter J A 2008 J.Alloys Compd.450 293
[13]Naik M,Rather S,Zacharia R,So C S,Hwang S,Kim A R,Nahm K S 2009 J.Alloys Compd.471 16
[14]Marashdeh A,Olsen R A,Lovvik O M,Kroes G J 2006 Chem. Phys.Lett.426 180
[15]Araújo C M,Ahuja R,Guillén J M 2005 Appl.Phys.Lett.86 251913
[16]Li S,Jena P,Ahuja R 2006 Phys.Rev.B 73 214107
[17]Kohn W,Sham L J 1965 Phys.Rev.140 1133
[18]Perdew J P,Burke K,Ernzerhof M 1996 Phys.Rev.Lett.77 3865
[19]Bloechl P E 1994 Phys.Rev.B 50 17953
[20]Kresse G,Furthmuller J 1996 Phys.Rev.B 54 11169
[21]Kresse G,Furthmuller J 1996 Comput.Mater.Sci.6 15
[22]Lauher J W,Dougherty D,Herley P J 1979 Acta Crystallogr.B 35 1454
[24]Brinks H W,Hauback B C 2006 J.Alloys Compd.354 143
[25]Kittel C 1995 Introduction to Solid State Physics(New York: Wiley)p7
[26]Silvera F I 1980 Rev.Mod.Phys.52 393
PACC:7115M,7125
*Project supported by the National Natural Science Foundation of China(Grant Nos.10804095,10664006),the Basic Applied Research Foundation of Yunnan Province,China(Grant Nos.2008CD062,2007A0017Z),the Key Science and Engineering Foundation of Yunnan University,China(Grant No.2007Z003B)and the Outstanding Youth Teachers Training Program of Yunnan University,China.
†Corresponding author.E-mail:yhe@ynu.edu.cn
Influence of Zr catalyst on reversible hydrogen storage characteristics of NaAlH4and Na3AlH6*
Ye Jia-Yu Liu Ya-Li Wang Jing-Lin He Yao†
(Department of Physics,Yunnan University,Kunming650091,China)
(Received 28 June 2009;revised manuscript received 20 November 2009)
The cell parameters,electron localization function and density of states of pure and Zr-doped NaAlH4and Na3AlH6are investigated using plane-wave pseudo-potential method based on density functional theory.The results show that NaAlH4and Na3AlH6are insulators characterized by a band gap of 4.6 and 3.1 eV,respectively.The Al and H atoms form covalent bonds and the Na and H atoms form ionic bonds in NaAlH4and Na3AlH6.When Zr replaces Na,the interaction between Zr and H is stronger than the primary Na—H bond,and the interaction between Al and H becomes weaker;when Zr replaces Al,the bond between Zr and H is weaker than the primary Al—H bond.Our calculations indicate that Zr-doped NaAlH4and Na3AlH6are more stable than that of the pure alanates,and the energy to remove H atom is significantly decreased.
hydrogen storage,NaAlH4,Na3AlH6,Zr-doped
book=229,ebook=229
*国家自然科学基金(批准号:10804095,10664006)、云南省应用基础研究基金(批准号:2008CD062,2007A0017Z)、云南大学理工科重点科研基金(批准号:2007Z003B)和云南大学中青年骨干教师培养计划资助的课题.
†通讯联系人.E-mail:yhe@ynu.edu.cn
