核磁共振内标法测定维生素C片中维生素C的含量
2010-08-07陈春丽田兰马晓丽孟磊李新霞新疆医科大学药学院分析测试中心乌鲁木齐市830054
陈春丽,田兰,马晓丽,孟磊,李新霞(新疆医科大学药学院分析测试中心,乌鲁木齐市830054)
维生素C作为维持机体正常生理功能的重要维生素之一,对人体健康有着重要的意义[1]。目前测定维生素C含量的方法很多,主要有滴定分析法、分光光度法、电极法、薄层色谱法、高效液相色谱法等。其中,滴定法操作简单,但是滴定终点难以准确判断;分光光度法操作费时;高效液相色谱法灵敏度高、选择性好,但前期处理较复杂,定量时需要高纯度物质作对照。
药物的核磁共振定量分析在我国已有多篇报道[2~6],美国、英国、欧洲等国和地区药典中都已先后引入了核磁共振定量分析方法[7,8],但有关维生素的相关研究较少。笔者采用核磁共振内标法对维生素C片中维生素C含量进行分析,以期建立核磁共振定量分析维生素C的方法。
1 仪器与试药
Varian unity Inova 600 MHz超导核磁共振波谱仪(美国Varian公司);Mettler toledo AB135-S型分析天平(瑞士Mettler toledo公司);氘代DMSO(美国CIL公司生产,D2O氘代度>99.8%,DMSO氘代度>99.9%);苯(天津市福晨化学试剂厂,分析纯);维生素C对照品(Sigma公司);维生素C片(成都锦华药业有限责任公司,批号:070505、070524、070613)。
2 方法与结果
2.1 试验条件
谱仪测定温度:21℃;观察频率:599.951 MHz;谱宽:9599.2 Hz;脉冲宽度(pw):11.65 μs;采样时间(at):2 s;延迟时间(d1):5 s;采样次数(nt):32次。
2.2 供试品溶液配制
2.2.1 优化仪器参数时的样品配置:精密称取维生素C对照品适量,置于5 mm核磁管中,并加入一定体积的内标苯及适量的溶剂氘代DMSO,即可进行1H-NMR谱检测。
2.2.2 定量分析时的样品处理方法:精确称取研磨后的维生素C片粉末适量,置于5 mm核磁管中,并加入一定体积的内标苯及适量的溶剂氘代DMSO,超声15 min,使其充分溶解,即得待测溶液。
2.3 谱仪操作方法
将装有待测样品的核磁管放入谱仪中,调整仪器参数,经认真调谐、匀场、采样得到1H-NMR谱,再进行谱图的相位和基线调整,对样品和内标的定量峰分别进行5次积分,取其平均值,得到积分结果。
2.4 溶剂和内标的选择
维生素C具有烯醇式己糖内酯立体结构,为水溶性化合物,在水中易溶解,但溶剂为D2O时,内标较难确定。本研究选用氘代DMSO为溶剂,苯为内标,其化学式和1H-NMR谱见图1。
由图1可见,样品、溶剂、内标峰完全分离,表明三者未发生化学反应和产生缔合。1H-NMR谱峰归属如下:δ11.03 ppm、δ8.31 ppm分别对应羟基1-H、2-H质子信号,δ7.37 ppm为内标苯的质子信号,δ4.85 ppm为羟基3-H、4-H合并在一起的质子信号,δ4.72 ppm、δ3.72 ppm分别为次甲基5-H、6-H质子信号,δ3.41 ppm为亚甲基-CH2-的质子信号,δ2.51 ppm为溶剂氘代DMSO的信号峰。

图1 以苯为内标时维生素C片在DMSO中的1H-NMR谱Fig 11H-NMR spectra of vitamin C with benzene as internal standard in DMSO
2.5 定量峰的选择
待测药片的1H-NMR谱见图2。由于维生素C片中除了主药外添加了其它辅料,导致谱图中出现干扰峰,但化学位移为δ4.72 ppm处的双峰、δ3.72 ppm处的多重峰受辅料影响不大,因此本试验考虑选择这2种峰作为样品定量峰,积分面积分别为As1和As2。在氢谱中,苯只有一单峰,即为内标定量峰面积积分为Ar,化学位移为δ7.37 ppm。
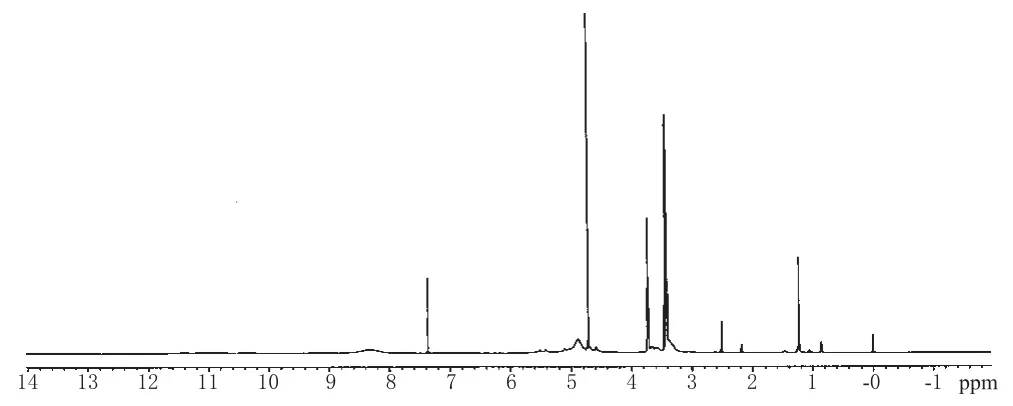
图2 维生素C片的1H-NMR谱Fig 21H-NMR spectra of vitamin C tablets
2.6 仪器参数的优化方法
2.6.1 pw的选择:不同pw下的As/Ar值见表1。其他仪器参数:nt=16次,d1=4 s,at=1 s。
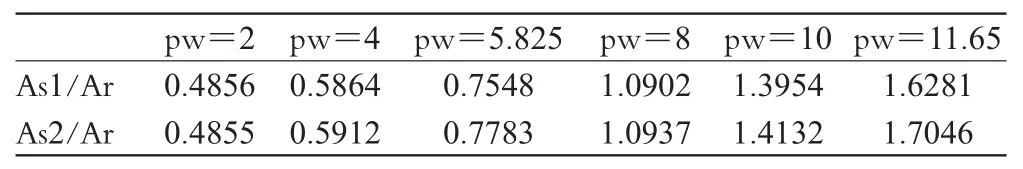
表1 不同pw对相对峰面积比的影响Tab 1Ratio of pw to As/Ar
由表1可见,随着pw的增加,样品定量峰与内标定量峰的积分面积比值随之增大,因此本试验选取pw=11.65 μs。
2.6.2 at的选择:不同at下的As/Ar值见表2。其他仪器参数:nt=16次,d1=4 s,pw=11.65 μs。

表2 不同at对相对峰面积比的影响Tab 2Ratio of at to As/Ar
由表2可见,当at=1~2 s时,所对应的As/Ar值较大,表明FID已衰减完全,故本试验选取at=2 s.
2.6.3 d1的选择:不同d1下的As/Ar值见表3。其他仪器参数:nt=16次,at=2 s,pw=11.65 μs。
由表3可见,当d1=5 s时,As/Ar最大,因此本试验选取d1=5 s。

表3 不同d1对相对峰面积比的影响Tab 3Ratio of d1 to As/Ar
2.6.4 nt的选择:测定不同nt下的As/Ar值见表4。其他仪器参数:d1=5 s,at=2 s,pw=11.65 μs。

表4 不同nt对相对峰面积比的影响Tab 4Ratio of nt to As/Ar
由表4可见,As/Ar随nt增大而增大,nt>32次后增加趋势不明显,为了节省试验时间、加快分析速度,本试验选取nt=32次。
2.7 1H-NMR谱内标法测定维生素C
2.7.1 定量方法:从1H-NMR谱中读取样品定量峰和内标定量峰积分面积,并用以下公式计算样品的含量:
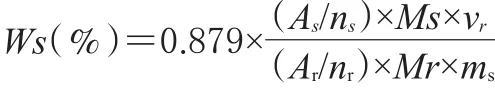
其中:As和Ar分别为被测样品和内标物质选定定量峰的峰面积;ns和nr分别为被测样品和内标物质定量峰包含的质子数;Ms和Mr分别为被测样品和内标的摩尔质量;vr为内标的体积。
2.7.2 含量测定。制备含有不同质量药品(批号:070505)的待测样品,分别测定1H-NMR谱,结果见表5。
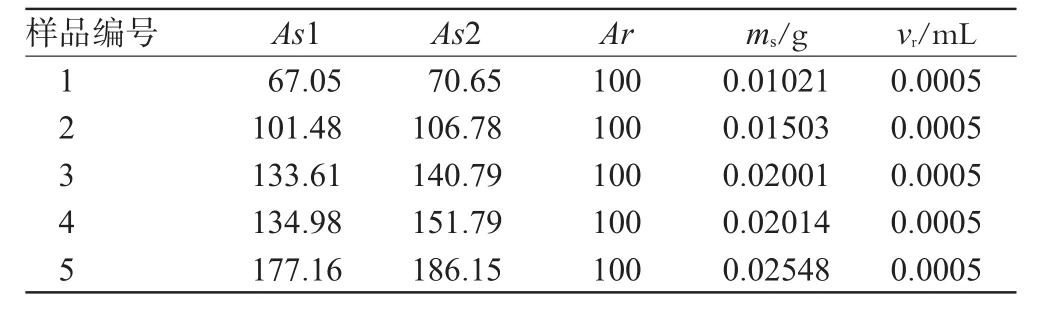
表5 维生素C含量的核磁共振数据Tab 5The data of the content of vitamin C
由化学位移为δ4.72 ppm处信号峰峰面积As1计算结果为:5份样品中维生素C的含量分别为39.1%、40.2%、39.8%、39.9%、41.4%,平均含量为40.1%,RSD=1.38%。
由化学位移为δ3.72 ppm处信号峰峰面积As2计算结果为:5份样品中维生素C的含量分别为41.2%、42.3%、41.9%、44.8%、43.5%,平均含量为42.74%,RSD=3.54%。
2.7.3 重复性试验。取3号样品,在同一试验条件下连续做5个1H-NMR谱,并计算维生素C与内标的定量峰峰面积比值以考察试验的重复性。结果,δ4.72 ppm、δ3.72 ppm处定量峰计算所得RSD分别为0.29%和0.76%,表明重复性较好。
2.7.4 回收率试验。分别在2、3、4、5号样中加入维生素C对照品8 mg,振荡使其充分溶解后测定1H-NMR谱,定量峰确定为δ4.72 ppm处信号,计算回收率,平均值为100.9%。加样回收率试验数据见表6。
批号为070524、070613的样品含量测定方法同上(定量峰δ4.72 ppm处为双峰),维生素C含量分别为43.6%、42.9%。
3 讨论
由于核磁共振定量分析是以结构分析为基础的分析方法,结构(包括构型和构象)分析、定性分析、定量分析可同步完成,方法自身具有简单、准确、专一性高和不破坏样品等优点。
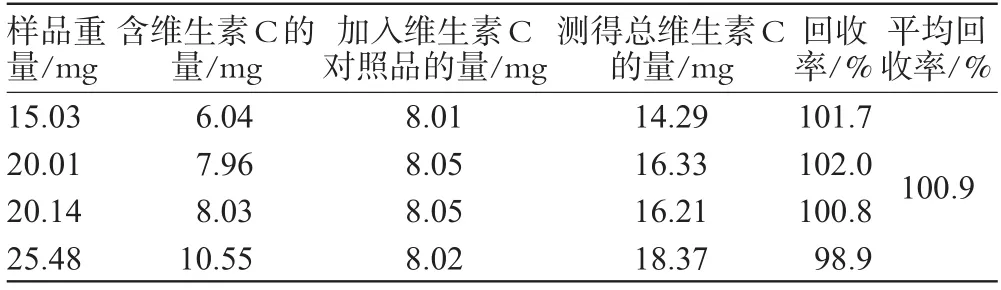
表6 加样回收率试验数据Tab 6Results of recovery test
1H-NMR谱法中内标的作用不同于常规方法中内标的作用。常规分析方法中,内标的作用是减少最终带入结果的操作误差,而在此方法中,内标是用以定量的依据。本研究曾选择2,2,3,3-三甲基甲硅烷基丙酸(TSP)作为内标,加入TSP后维生素C中的次甲基峰δ7.36 ppm位移至δ4.80 ppm,几乎与水峰重叠。由此可知,内标TSP与重水发生了缔合,不适合作定量分析。由1H-NMR谱内标法与高效液相定量分析方法相比,样品用量小,前处理方法简单,一般无需分离,定量时无需化合物的纯品作为对照标准,可以对无对照品的药品进行含量分析和质量控制,结构鉴定和定量分析可同步完成,分析速度快。足够的at可以保证自由感应衰减信号衰减完全,从而提高分辨率。而合适的d1可保证谱图强度不会被饱和,有利于对谱峰进行正确的积分。在仪器测试状态调整到最优时,该法准确度和精密度可以接近高效液相色谱法。由本试验数据可知,该法简便、准确,可以用于维生素C的含量分析,也可为后续研究核磁共振定量分析其它药物提供参考。
[1]张石革,孙定人.维生素缺乏症与维生素的补充[J].中国药房,2002,13(6):382.
[2]王强,汪茂田,张志权.核磁共振法定量测定替米考星含量[J].分析测试学报,2003,22(6):101.
[3]刘英,胡昌勤.核磁共振在抗生素药物定量分析中的应用[J].药物分析杂志,2001,21(6):447.
[4]沈文斌,王庆峰,廖海平.药物对映体含量测定的核磁共振法研究Ⅰ.非洛地平[J].药物分析杂志,1996,16(4):230.
[5]沈文斌,张灿,王文庆.四氢小檗碱对映体的核磁共振研究[J].中国药科大学学报,1997,28(3):132.
[6]王庆峰,杭太俊,张正行.核磁共振法测定萘普生对映体相对含量[J].中国药科大学学报,1999,30(1):31.
[7]United States Pharmacopoeia Commission.USP24/NF19:1221~1222.
[8]United States Pharmacopoeia Commission.USP24/NF19:1959~1965.
