高效液相色谱法测定硫酸卷曲霉素的有关物质
2010-07-29陈汝红张艳艳
陈汝红,孙 婷,张艳艳
(1.河北省药品检验所,河北 石家庄 050011; 2.河北省唐山市药品检验所,河北 唐山 063000)
硫酸卷曲霉素为多肽类抗生素,对结核分枝杆菌有抑制作用,是用于肺结核病的二线治疗药物。它含有4个极相近的组分(ⅠA,ⅠB,ⅡA,ⅡB),其中卷曲霉素I为主要活性物质,体外活性比卷曲霉素Ⅱ高数倍。ChP2005,USP30,BP2002均涉及卷曲霉素Ⅰ的含量控制,但未见硫酸卷曲霉素有关物质测定的文献报道。笔者参考Chp2005[1],USP30,BP2002建立了其有关物质的测定方法,可用于硫酸卷曲霉素及其粉针剂的质量控制。
1 仪器及试药
Waters 2695型高效液相色谱仪,Waters 2487型紫外检测器。卷曲霉素标准品(中国药品生物制品检定所,批号为0339-9802);硫酸卷曲霉素原料1批(南宁中科药业有限责任公司,批号为0604101);注射用硫酸卷曲霉素4批(1批由华北制药股份有限公司提供,规格为0.5 g,批号为051201;3批由南宁中科药业有限责任公司提供,规格为 0.5 g,批号分别为 0404001,0506001,0601001)。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:Gemini C18柱 (150 mm ×4.6 mm,5μm);流动相:0.016 mol/L己烷磺酸钠溶液-甲醇-乙腈-冰醋酸(60∶30∶8∶2);检测波长:268 nm;进样量:20 μL。取卷曲霉素标准品适量,用水制成0.5 mg/mL的溶液,进样测定,高效液相色谱图见图1。卷曲霉素Ⅱ峰对卷曲霉素Ⅰ峰的相对保留时间约为0.45,卷曲霉素Ⅰ峰和卷曲霉素Ⅱ峰分别与相邻杂质峰的分离度均大于1.5。
2.2 供试品溶液制备
取供试品适量,用水溶解并制成1.0 mg/mL的溶液,作为供试品溶液。
2.3 专属性考察
对硫酸卷曲霉素原料进行破坏试验。取原料50 mg,置50 mL量瓶中,加1 mol/L氢氧化钠溶液5.0 mL,放置10 min,加1 mol/L盐酸溶液5.0 mL中和,加水稀释至刻度,摇匀(碱破坏)。取原料50 mg,置50 mL量瓶中,加 3 mol/L盐酸溶液 5.0 mL,放置10 min,加3 mol/L氢氧化钠溶液5.0 mL中和,加水稀释至刻度,摇匀(酸破坏)。分别取上述两种破坏溶液进样测定。结果见图2和图3。可见,卷曲霉素Ⅰ峰、卷曲霉素Ⅱ峰分别与降解产物峰有较好的分离。
2.4 检测限确定
取标定的硫酸卷曲霉素对照品(南宁中科药业有限责任公司提供,批号为0604101,含卷曲霉素Ⅰ按卷曲霉素ⅠB计算为68.6%)适量,用水溶解并制成系列质量浓度的溶液,进样测定。以信噪比 S/N为3时,注入仪器的量确定卷曲霉素Ⅰ的检测限为2 ng。
2.5 样品测定
分别取上述标准品1批、原料1批、制剂4批适量,用水制成1.0 mg/mL的溶液,取20 μL注入液相色谱仪,记录色谱图至卷曲霉素Ⅰ峰保留时间的2倍。色谱图见图4。按峰面积归一法计算卷曲霉素Ⅰ、卷曲霉素Ⅱ以及杂质的含量,结果见表1。
2.6 耐用性试验
采用另外两个不同品牌的色谱柱,测定以上6批样品的有关物质,结果基本一致。
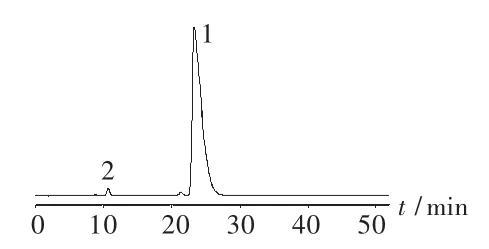
图1 系统适用性试验图谱
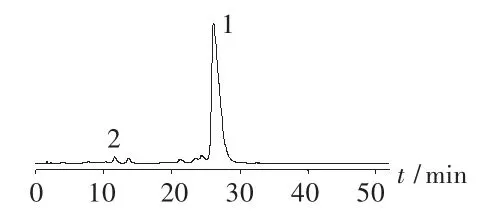
图2 碱破坏试验图谱

图3 酸破坏试验图谱

图4 样品测定图谱

表1 硫酸卷曲霉素有关物质测定结果(%)
3 讨论
卷曲霉素为碱性水溶性抗生素,含有多个极相近的组分,有关物质不易分离。在ChP2005中,采用离子对色谱法,用C18柱对卷曲霉素Ⅰ、卷曲霉素Ⅱ进行分离测定,卷曲霉素Ⅰ峰与卷曲霉素Ⅱ峰之间分离度较小,不利于有关物质的检出。本试验参考ChP2005卷曲霉素Ⅰ的测定方法,对甲醇及乙腈的比例、离子对试剂的浓度、流动相的pH进行了优化。在所选色谱条件下,在卷曲霉素Ⅰ相对保留时间0.9的位置分离得到1个2%左右的未知杂质,分离度满意,卷曲霉素Ⅱ与其相邻杂质峰的分离度亦符合药典要求。
取卷曲霉素标准品适量,用水溶解并稀释成适当质量浓度的溶液,在200~350 nm波长范围内扫描。结果卷曲霉素在268 nm波长处有最大吸收,因此将测定波长定为268 nm。
为了有效检出有关物质,将测定质量浓度定为1.0 mg/mL,进样20 μL可检出0.01%的杂质(按卷曲霉素ⅠB计算),能满足杂质控制要求。
USP29及BP2002采用氰基柱,将卷曲霉素Ⅰ分离成ⅠA和ⅠB两个峰,其他峰未经确证。英美药典方法采用的氰基柱及流动相缓冲盐不易得,而且不须控制ⅠA和ⅠB的组成比例,笔者仅改进了ChP2005卷曲霉素Ⅰ的测定方法,改进后的方法检出的有关物质数目较多,杂质总和与用英美药典方法测定的相近。
[1]国家药典委员会.中华人民共和国药典(二部)[M].北京:化学工业出版社,2005:743.
