Acinar cell carcinoma of the pancreas in a young patient with chronic pancreatitis
2010-06-29FatimaZahraKebirAhlemLahmarNafaaArfaSaberManaiMohamedAliElOuaerSaadiaBouraouiCaroleGouttalierandSabahMezabiRegaya
Fatima-Zahra Kebir, Ahlem Lahmar, Nafaa Arfa, Saber Manai, Mohamed Ali El Ouaer, Saadia Bouraoui, Carole Gouttalier and Sabah Mezabi-Regaya
Sidi Daoued La Marsa, Tunisia
Acinar cell carcinoma of the pancreas in a young patient with chronic pancreatitis
Fatima-Zahra Kebir, Ahlem Lahmar, Nafaa Arfa, Saber Manai, Mohamed Ali El Ouaer, Saadia Bouraoui, Carole Gouttalier and Sabah Mezabi-Regaya
Sidi Daoued La Marsa, Tunisia
BACKGROUND:Acinar cell carcinoma (ACC) is a rare malignancy of the pancreas arising from acinar cells. Unlike ductal adenocarcinoma, this tumor rarely presents with pancreatitis.
METHODS:We present a case of ACC associated with chronic calcifying pancreatitis, and a review of the literature focusing on diagnosis and management.
RESULTS:A 43-year-old man was proposed for Wirsungojejunal derivation for chronic pancreatitis. Histopathological examination of the tissue extracted revealed an ACC. Duodenopancreatectomy was performed. Six months postoperatively, the patient developed hepatic metastasis and was treated with gemcitabine as palliative chemotherapy.
CONCLUSIONS:The clinical presentation of ACC of the pancreas is not specific and the tumor can be underdiagnosed when associated with chronic pancreatitis. Data regarding course, treatment, and prognosis of this tumor are generally lacking.
(Hepatobiliary Pancreat Dis Int 2010; 9: 103-106)
pancreas; acinar cell carcinoma; pancreatitis
Introduction
Acinar cell carcinoma (ACC) of the pancreas is a malignant tumor arising from acinar cells. It is a rare tumor accounting for approximately 1% of pancreatic exocrine tumors. Clinical presentation is not specific and this tumor rarely presents with pancreatitis.[1]Treatment protocols and prognosis are generally lacking. The tumor arising from acinar cells generally carries a better prognosis than ductal adenocarcinoma. We report a 43-year-old man with chronic pancreatitis who was found to have ACC.
Case report
A 43-year-old man was admitted to the surgical unit of this hospital for 2 months of epigastric abdominal pain, which was moderate in severity and radiated to the back without other symptoms. Physical examination showed moderate epigastric tenderness. His medical history showed many attacks of pancreatitis in 2 previous years. He had a history of drinking, and occasional smoking stopped 13 years ago. He had no family history of pancreatitis or gastrointestinal or pancreatic diseases, but was followed up for chronic calcifying pancreatitis. Initial laboratory evaluation showed nothing abnormal.
Ultrasound showed a significant dilatation of Wirsung's duct with many stones and calcifications in the pancreatic parenchyma. CT scan confirmed the same lesions (Fig. 1). No tumoral masses were observed in the head of the pancreas and nor dilatation of the common bile duct and cholelithiasis. The gallbladder and liver were normal. Chronic calcifying pancreatitis was suspected.
The patient was taken to the operating room for Wirsung's-jejunal derivation and extraction of pancreatic stones (Frey procedure). It consists on excision of head pancreatic tissue with stones ablation and latero-lateral Wirsung's jejunal unastomosis.
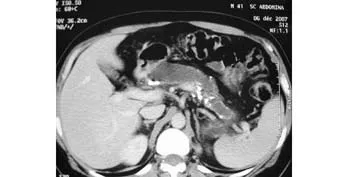
Fig. 1. CT scan showing significnt dilatation of Wirsung's duct with many stones and calcifications of the pancreatic parenchyma.
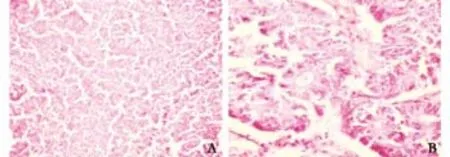
Fig. 2. Light microscopic appearance. A: At low power (original magnification ×250), acinar pattern of the tumor with neoplastic cells arranged in small glandular units, giving a cribriform appearance. Within the tumor are cell islands. There is an abundant fine microvasculature.B:At high power (original magnification ×400), an abundant pink, eosinophilic cytoplasm with granules. The nuclei, round to oval, are relatively uniform.
Histologic examination of the tissue extracted with the stones revealed malignant, solid, uniform sheets of cells with a homogeneous appearance. At higher power, an abundant pink, eosinophilic cytoplasm with granules was visible without highly pleomorphic nuclei (Fig. 2). This appearance was characteristic of ACC. The diagnosis was confirmed by d-PAS staining, which highlighted the cytoplasmic granules in tumor cells. Immunohistochemical staining of the tumor cells was negative for neuron-specific enolase (NSE), chromogranin and synaptophysin.
Four weeks later, cephalic duodenopancreatectomy was performed.
Macroscopic examination showed a grossly fleshy tumor of 4×4 cm that spread to the neighboring intestinal resection segment. The pancreas was hypertrophic and, hard on section, with multiple stones in the dilated ducts (Fig. 3).
The diagnosis of ACC was confirmed histologically. Regional lymph nodes were negative in the resected specimen.
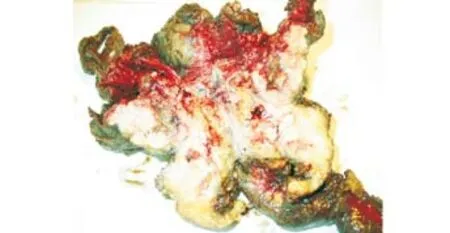
Fig. 3. A flesh tumor of 4×4 cm spreading to the neighboring intestinal anastomosing segment. The pancreas is hard on section with multiple stones in the dilated ducts.
Adjuvant chemotherapy or radiotherapy was not given. The patient developed hepatic metastasis 6 months after operation, and he was treated with gemcitabine as palliative chemotherapy.
Discussion
Acinar cell carcinoma is a pancreatic epithelial tumor that can occasionally have a neuroendocrine component. It is an uncommon tumor, representing only 1%-2% of all exocrine pancreatic tumors.[1]There are very limited reports on this tumor, its clinical-pathological features treatment outcome.
ACC predominantly affects white elderly men at mean age of 62 years. Only few cases are found in children.
Most cases of ACC are asymptomatic. Clinical presentations include weight loss (50%), abdominal pain (32%), nausea and vomiting (20%), and elevated lipase levels (16%).[2]Jaundice is not common even when tumor involves the head of the pancreas. Functional ACC can present with several characteristic clinical syndromes. Patients with elevated lipase levels may have systemic manifestations of subcutaneous fat necrosis. Endocrine manifestations are specific but uncommon features of ACC. Hypoglycemia occurs secondary to tumor secretion of insulin, insulin-like growth factors, and glucagon.[3]Our case was unusual because it did not match the average age or clinical profile of ACC.
ACC of the pancreas can be stained by several immunohistochemical markers. It can be associated with increased levels of tumor markers such as α-fetoprotein and carcinoembryonic antigen.[4]In our case, none of these markers were found.
There are rare studies on imaging features of ACC, which is ACC well circumscribed, sizable, and hypodense. ACC is often heterogenous, with occasionalinternal calcifications and/or tumoral bleeding. MRI may be helpful in showing tumor thrombosis in cases of splenic or portal vein thrombosis.[5,6]
Definitive diagnosis of ACC requires histopathological analysis. ACC involves the head or the tail of the pancreas (56% and 36%, respectively). Large tumors tend to have multiple areas of hemorrhage and necrosis.[1]Cystic changes can be seen but not prominently.[7,8]
Microscopically, ACC is encapsulated and highly cellular with stroma and without stromal desmoplasia. Focal capsular invasion is seen in most cases, with finger-like projections extending into adjacent pancreatic parenchyma. Vascular invasion occurs in 2/3 of patients and perineural invasion in 32% of cases. Thick fibrous bands separate the tumor cells into lobules.[9]The tumor has four basic patterns of growth: acinar, cellular, trabecular and glandular. The acinar pattern is the most common but often mixed with others.
A variant known as acinar cystadenocarcinoma is predominantly cystic; another variant is described as mixed acinar-endocrine tumor.
ACC has hyperchromatic round/oval nuclei with prominent, single nucleoli. The cytoplasm is abundant, eosinophilic, and granular due to zymogen granules that produce exocrine enzymes (trypsin, lipase and chymotrypsin). The diagnosis of ACC is based on the presence of zymogen granules, which stain positive with periodic acid-Schiff and are diastase resistant. In wellgranulated cases, this staining pattern is supportive of an ACC diagnosis.[1,9]
More sensitive and specific methods use immunohistochemical assays to rule out other neuroendocrine tumors. These detect specific enzymes (trypsin, chymotrypsin, amylase and lipase), which are highly expressed in almost all ACCs.
No studies have been conducted to investigate the relationship between the presence of different immunohistochemical markers and prognosis. Though there are several immunohistochemical markers for ACC of the pancreas, none has proven to be of value for early detection.[3]
Because of the rarity of this neoplasm, its therapeutic approach is still not codified. Treatment choice depends on the stage of the disease at time of diagnosis. Surgical resection is the best treatment in the absence of distant metastasis. The extent on resection depends on the extent of the tumor. En bloc resection has been reported in patients with locally advanced disease.[1]Surgical resection offers the only chance of cure, and the resectability rate is 64%, much higher than that for ductal adenocarcinoma.[10,11]
Adjuvant chemotherapy and radiotherapy may help to prolong survival.[11]Intraperitoneal chemotherapy with cisplatin has been used for peritoneal metastasis, and 5-Fluorouracil is the most common chemotherapeutic agent. In our case, the tumor was removed by en bloc resection, but not given adjuvant chemotherapy or radiation therapy.[10-12]
The prognosis of ACC is poor. The mean survival of patients with ACC is approximately 18 months. The 3-year survival is 26% and the 5-year survival is only 6%. Cancer of the pancreas in Japan and the US showed the same results in a large series of 762 and 115 patients.[9,12]The prognosis of these tumors was better than that of ductal carcinomas but worse than that of islet cell tumors. Half of the patients has distant metastasis at the time of diagnosis, and an additional 25% developed metastasis later. Although unreported in this particular type of tumor, pancreatitis secondary to other pancreatic tumors is well known. One study suggests that pancreatic carcinoma may present as acute pancreatitis in up to 13% of cases and wherever pancreatic carcinoma is associated with pancreatitis, the diagnosis is often delayed.[13]This may be the case when the tumor co-exists with gallstones, providing an alternative cause of pancreatitis.[13,14]Predictors of poor prognosis include age greater than 60 years, elevated serum lipase, and tumors larger than 10 cm in diameter.[15-19]
Funding:None.
Ethical approval:Not needed.
Contributors:KFZ wrote the first draft of this case report. All authors contributed to the intellectual context and approved the final version. AN is the guarantor.
Competing interest:No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
1 Klimstra DS, Rosai J, Heffess CS. Mixed acinar-endocrine carcinomas of the pancreas. Am J Surg Pathol 1994;18:765-778.
2 Ordónez NG, Mackay B. Acinar cell carcinoma of the pancreas. Ultrastruct Pathol 2000;24:227-241.
3 Chen CP, Chao Y, Li CP, Lee RC, Tsay SH, Chi KH, et al. Concurrent chemoradiation is effective in the treatment of alpha-fetoprotein-producing acinar cell carcinoma of the pancreas: report of a case. Pancreas 2001;22:326-329.
4 Lin YC, Lee PH, Yao YT, Hsiao JK, Sheu JC, Chen CH. Alphafetoprotein-producing pancreatic acinar cell carcinoma. J Formos Med Assoc 2007;106:669-672.
5 Tatli S, Mortele KJ, Levy AD, Glickman JN, Ros PR, Banks PA, et al. CT and MRI features of pure acinar cell carcinoma of the pancreas in adults. AJR Am J Roentgenol 2005;184:511-519.
6 Khalili M, Wax BN, Reed WP, Schuss A, Drexler S, Weston SR, et al. Radiology-pathology conference. Acinar cell carcinoma of the pancreas. Clin Imaging 2006;30:343-346.
7 Stamm B, Burger H, Hollinger A. Acinar cell cystadenocarcinoma of the pancreas. Cancer 1987;60:2542-2547.
8 Virlos I, Papazachariou I, Wiliamson R. Acinar cell carcinoma of the pancreas with and without endocrine differentiation. HPB (Oxford) 2002;4:87-90.
9 Wisnoski NC, Townsend CM Jr, Nealon WH, Freeman JL, Riall TS. 672 patients with acinar cell carcinoma of the pancreas: a population-based comparison to pancreatic adenocarcinoma. Surgery 2008;144:141-148.
10 Holen KD, Klimstra DS, Hummer A, Gonen M, Conlon K, Brennan M, et al. Clinical characteristics and outcomes from an institutional series of acinar cell carcinoma of the pancreas and related tumors. J Clin Oncol 2002;20:4673-4678.
11 Antoine M, Khitrik-Palchuk M, Saif MW. Long-term survival in a patient with acinar cell carcinoma of pancreas. A case report and review of literature. JOP 2007;8:783-789.
12 Kitagami H, Kondo S, Hirano S, Kawakami H, Egawa S, Tanaka M. Acinar cell carcinoma of the pancreas: clinical analysis of 115 patients from Pancreatic Cancer Registry of Japan Pancreas Society. Pancreas 2007;35:42-46.
13 Kohler H, Lankisch PG. Acute pancreatitis and hyperamylasaemia in pancreatic carcinoma. Pancreas 1987;2:117-119.
14 Gambill EE. Pancreatitis associated with pancreatic carcinoma: a study of 26 cases. Mayo Clin Proc 1971;46:174-177.
15 Aqel B, Scolapio J, Nguyen J, Krishna M, Raimondo M. Recurrent pancreatitis due to a cystic pancreatic tumor: a rare presentation of acinar cell carcinoma. JOP 2004;5:151-154.
16 Thomas P, Nash G, Aldridge M. Pancreatic acinar cell carcinoma presenting as acute pancreatitis. HPB (Oxford) 2003;5:111-113.
17 Rouleau C, Serre I, Roger P, Guibal MP, Galifer RB, Bonardet A, et al. Acinar cell carcinoma of the pancreas in a young patient with cells immunoreactive for somatostatin. Histopathology 2006;48:307-309.
18 Riechelmann RP, Hoff PM, Moron RA, da Câmera Lopes LH, Buzaid AC. Acinar cell carcinoma of the pancreas. Int J Gastrointest Cancer 2003;34:67-72.
19 Colombo P, Arizzi C, Roncalli M. Acinar cell cystadenocarcinoma of the pancreas: report of rare case and review of the literature. Hum Pathol 2004;35:1568-1571.
December 12, 2008
Accepted after revision August 13, 2009
Author Affiliations: Department of Pathology (Kebir FZ, Lahmar A, Bouraoui S, Gouttalier C and Mezabi-Regaya S) and Department of Surgery (Arfa N, Manai S and Ouaer MAE), Mongi Slim Hospital, 2046 Sidi Daoued La Marsa, Tunisia
Nafaa Arfa, MD, Department of Surgery, Mongi Slim Hospital, 2046 Sidi Daoued La Marsa, Tunisia (Email: nafaa.arfa@rns.tn) © 2010, Hepatobiliary Pancreat Dis Int. All rights reserved.
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Clinical management of hepatitis B virus infection correlated with liver transplantation
- Is bactibilia a predictor of poor outcome of pancreaticoduodenectomy?
- Oxidative stress and lipid peroxidation products: effect of pinealectomy or exogenous melatonin injections on biomarkers of tissue damage during acute pancreatitis
- Letters to the Editor
- Simplifying living donor liver transplantation
- Meetings and Courses