TSA对食管癌细胞系EC9706细胞周期作用及分子机制的探讨
2010-06-15马俊芬赵继敏杨洪艳黄幼田董子明
刘 霞,冯 婷,马俊芬,赵继敏,杨洪艳,黄幼田,董子明
(郑州大学基础医学院病理生理学教研室 450052)
组蛋白乙酰化酶(histoneacetylase,HAT)和组蛋白去乙酰化酶(histonedeacetylase,HDAC)是调节组蛋白乙酰化状态的一对功能相互拮抗的蛋白酶,它们相互的动态平衡控制着染色质的结构和基因的表达。曲古菌素A(trichostatin A,TSA)是一种组蛋白去乙酰化酶(histonedeacetylase,HDACs)抑制剂,能够抑制HDACs的活性。由于它能够改变组蛋白的乙酰化状态,从而可以选择性地调控肿瘤相关基因。近年来,研究提示去乙酰化转移酶抑制剂可明显抑制肿瘤的生长,虽然目前对其作用机制仍不十分清楚,但其中一个重要机制是通过阻滞细胞周期来实现其细胞抑制作用的[1]。具体的机制因去乙酰化转移酶抑制剂药物和肿瘤的各异而不同。调控的相关基因也不相同。TSA对食管癌细胞周期影响的尚未见报道,因此,本研究主要观察TSA对食管癌EC9706细胞周期的影响,探讨TSA对EC9706细胞周期发挥作用的分子机制。
1 材料与方法
1.1 主要试剂和仪器 TSA购自Sigma(USA)公司,由DMSO溶解至1 mol/L,使用前按照相应浓度稀释加入培养基中。RPMI-1640购自Gibco BRL公司,胎牛血清购自天津TBD生物有限公司,P21、P27、CyclinD1鼠抗人单抗和抗CDK4多抗购自美国Santa Cruz;ECL显色试剂盒购自北京中杉生物工程公司,蛋白垂直电泳仪和印迹转膜仪购自美国Bio-Rad公司,流式细胞仪购自FACSsort,BD公司,食管癌细胞株 EC9706由郑州大学病理生理学教研室提供。
1.2 细胞培养 将EC9706细胞放在含10%胎牛血清、100 mg/mL链霉素、100 mg/mL青霉素的RPM I-1640培养液里,放在37℃,5%CO2培养箱中培养。取对数生长期的细胞处理后分组,实验组分为 0.5、1.0、1.5 μ mol/L TSA。对照组换为等浓度的DMSO同时培养24 h后收集。
1.3 TSA细胞生长抑制试验(MT T法) 实验分组:实验组,细胞分别给予 0.5、1.0、1.5 μ mol/L TSA 处理,使用不含胎牛血清的培养基;空白对照组M TT法检测不同浓度 TSA对细胞生长的抑制作用,细胞经0.05%胰酶消化处理制成3×104个/mL细胞悬液,接种在96孔板中,每孔加入100 μ L,每个浓度设6个复孔,以未加药细胞作为对照,待细胞贴壁后,分别加入 0.5、1.0、1.5 μ mol/L TSA 作用 24 h 后,加入 20 μL M TT(5 mg/mL)在5%CO2、37℃的恒温培养箱培养 4 h后弃上清液,每孔再加入150 μ L DMSO,待充分溶解后轻微摇床振荡10 min,上酶标仪检测,用波长570 nm比色测定OD值,计算抑制率。抑制率(%)=(空白对照组OD570-实验组OD570)/对照组OD570。
1.4 流式细胞仪检测细胞周期 取对数生长期的EC9706细胞,以无血清的RPMI-1640培养24 h同步后,按照上述分组作用24 h后,以PBS洗涤,用 70%的冷乙醇在4℃以下固定24 h以上,然后离心弃乙醇(3 000 r/min离心10 min),再加入1 mL PBS制成细胞悬液后加入 RnaseA酶(终浓度为 10 μ g/mL),室温放置30 min,再用 50 μ g/mL的碘化丙啶(PI)染色后,用流式细胞仪分析细胞周期。
1.5 Western Blot检测 p21、p27、CCND1、CDK4D 蛋白的表达 参照《分子克隆实验指南》提取肿瘤细胞总蛋白,测定浓度后,在标准蛋白曲线下算出50 μ g蛋白上样量,以 B-actin为内参照,Western Blot检测肿瘤细胞表面各基因的表达。将蛋白经SDS-PAGE后转移至PVDF膜,5%BSA摇床封闭3 h后加入相应的一抗,4℃冰箱孵育过夜,TBST洗涤3次,二抗孵育1 h后用化学发光法进行X线胶片曝光显影。用凝胶成像仪进行图像定量分析。
1.6 统计学方法 所得数据用SPSS10.0统计软件处理,多样本均数采用单因素方差分析。以P<0.05为差异有统计学意义。
2 结 果
2.1 TSA对EC9706细胞增殖影响 见表1,不同浓度TSA处理 EC9706细胞 24 h后,与空白对照组比较,0.5 μ mol/L TSA处理组细胞的抑制率差异无统计学意义,而1.0 μ mol/L TSA处理的细胞抑制率显著增加,并随着TSA作用浓度的升高而升高(P<0.05)。
表1 不同浓度的TSA对EC9706细胞增殖的影响(±s,n=6)

表1 不同浓度的TSA对EC9706细胞增殖的影响(±s,n=6)
与空白对照组比较,*:P<0.05;不同浓度TSA处理组间两两比较,#:P<0.05。
组别 抑制率(%)空白对照组 7.8±3.74 0.5 μ mol/L TSA 处理组 7.96±0.12#1.0 μ mol/L TSA 处理组 20.03±0.21*#1.5 μ mol/L TSA 处理组 26.40±0.58*#

图1 不同浓度TSA对EC9706细胞细胞周期的影响
2.2 TSA对EC9706细胞细胞周期的作用 见图 1,0.5、1.0、1.5 μ mol/L TSA 作用 EC9706细胞24 h 后,与空白对照组比较,0.5 μ mol/L TSA处理组细胞的周期无明显差异,而1.0 μ mol/L TSA处理后滞留在G1期的细胞显著增加,S期细胞明显减少,并随着TSA作用浓度的升高阻滞在G1期的细胞显著增高(P<0.05)。
2.3 EC9706 细胞 p21、p27、CCND1、CDK4D 的表达变化 见图 2,0.5、1.0、1.5 μ mol/L TSA 作用 EC9706 细胞 24 h 后,与空白对照组比较,0.5 μ mol/L TSA 处理组细胞 p21、p27、CCND1、CDK4D蛋白的表达差异无统计学意义,而 1.0 μ mol/L TSA处理的细胞p21、p27蛋白的表达显著提高,并随着TSA作用浓度的升高而增加(P<0.05),CCND1、CDK4D蛋白的表达显著下降,并随着TSA作用浓度的升高而降低(P<0.05)。
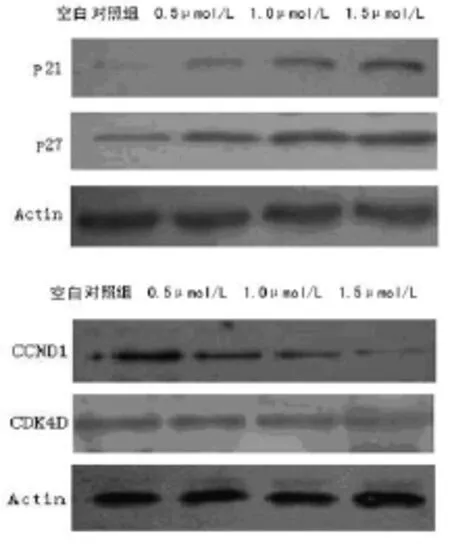
图 2 EC9706 细胞 p21、p27、CCND1、CDK4D 的表达变化
3 讨 论
组蛋白的乙酰化和去乙酰化主要是通过HAT和HDAC来实现的[2]。生理状态下,它们相互的动态平衡控制着染色质的结构和基因的表达。一旦平衡在异常状态下被破坏,HDAC活性增强,原有的基因表达失衡,调控细胞周期和细胞增殖的分子表达紊乱,从而导致细胞恶变。组蛋白去乙酰化抑制剂可以使某些部位的核心组蛋白高度乙酰化,导致某些基因的启动子激活,从而使与周期相关的蛋白得到表达,进而使肿瘤细胞出现增生停止、分化、凋亡转移抑制等变化。TSA是一种组蛋白去乙酰化酶抑制剂(HADCI),能特异地抑制组蛋白去乙酰化;使高度酰基化的组蛋白积累,改变染色体结构,抑制细胞增殖,诱导肿瘤细胞分化和(或)凋亡,是一类具有广泛应用前景的抗肿瘤药物之一[3]。有研究表明,TSA可以以时间和剂量依赖性方式显著抑制多种肿瘤细胞增殖,本研究证实了TSA可以抑制肿瘤细胞生长,可使食管癌细胞EC9706细胞周期停滞于G0/G1期。并且发现 0.5 μ mol/L TSA作用 EC9706细胞对细胞生长无明显影响,与空白对照组比较抑制率差异无统计学意义,而 1.0 μ mol/L TSA作用EC9706细胞显著抑制细胞生长,并且抑制率明显增加,并且表现出一定的剂量效应。进一步研究表明TSA对食管癌细胞周期的阻滞作用是通过调控一些细胞周期相关基因的表达来起作用的,TSA可以明显增加p21、p27等许多细胞周期抑制基因的表达,同时降低了CCND1、CDK4D等细胞周期促进基因的表达,并且随着TSA作用浓度的增加而增强。p21、p27是细胞周期调控中非常重要的基因,能够与CCND1形成复合物使细胞周期停滞在G1期;它还可以通过C端与PCNA相互作用,阻断PCNA活化DNA聚合酶的活性从而抑制DNA的合成,使细胞周期停滞[4-6]。其主要是抑制细胞周期,参与细胞的生长、分化、衰老及死亡过程,同时又与肿瘤发生密切相关,在细胞的生理、病理过程中发挥重要的作用。而CCND1过表达与某些肿瘤的组织类型相关,其功能主要是促进细胞增殖,是G1期细胞增殖信号的关键蛋白质,被视为癌基因,其过度表达可致细胞增殖失控而恶性化,但是它在不同肿瘤中阳性检出率不同,与组织分化程度相关,在分化差的肿瘤中表达增强。CDK则是一类重要的丝氨酸/苏氨酸蛋白激酶,包括CDK1~7种,主要生物学作用是启动DNA的复制和诱发细胞的有丝分裂[7-8],以复合物形式出现,在G1早期,CCND表达并与CDK2或CDK4结合,成为始动细胞周期的启动子[9-12],1.0 μ mol/L浓度的TSA对EC9706细胞有明显的生长抑制作用,并呈现一定的量效关系。本研究发现TSA在1.0 μ mol/L浓度之上可明显诱导食管癌细胞EC9706细胞周期阻滞,可明显增强EC9706细胞中p21、p27等基因的表达,明显降低 CCND1、CDK4D等基因的表达,但是否TSA抑制EC9706细胞的生长还与其它基因表达的改变相关,具体的机制有待继续研究探讨。
[1] Tian FQ,Chen XS,Xie J,et al.Effect of Trichostatin A on expression of HDAC1 in K562 cells[J].China Journal of Modern Medicine,2006,16(11):1642.
[2] And M,Hansen R,Ding RX,et a1.Disruption of 3D tissue integrity facilitates adenovirus infection by deregu lating the coxsackievirus and adenovirus receptor[J].Proc Natl Acad Sci USA,2003,100(4):1943.
[3] Seoj S,Chon Y,Kim R,et al.Cell cycle arrest and lyticinduction of EBV-transformed B lymphoblastoid cells by a his-tone deacetylase inhibitor,T richostatin A[J].Oncol Rep,2008,19(1):93.
[4] 张梅芳.原发性肝细胞癌组织中p53和p21 WAF/CIP1及MDM2蛋白的表达及其临床意义[J].中华肿瘤防治杂志,2007,14(15):1121.
[5] Radhakrishnan SK,Feficiano CS,Najmabadi F,et al.Constitutive expression of E2F-1 leads to p21-dependent cell cycle arrest in S phase of the cell cycle[J].Oncogene,2004,23(23):4173.
[6] 王振国,郭和清.膀胱癌 Ha-ras基因突变、p21蛋白表达及其意义[J].中国现代医学杂志,2003,13(8):59.
[7] Kalpana S,Joshi.In vitro antitumor properties of a novel cyclin-dependent kinase inhibitor,P276-00[J].Mol Cancer Ther,2007,6(3):918.
[8] Yu Q,Sicinska E,Geng Y,et al.Requirement for CDK4 kinase function in breast cancer[J].Cancer Cell,2006,9(1):23.
[9] Henkens T,Papeleu P,Elautl G,et al.T richostatin A,acritical factor in maintaining the functional differentiation of primary cultured rat hepatocytes[J].Toxicol Appl Pharmacol,2007,218(1):64.
[10]Sachs MD,Rauen KA,Ramamurthy M,et al.Integrin V and coxsackie adenovirusreceptor expression in clinical bladder[J].Cancer Urology,2002,60:531.
[11]Rauen KA,Sudilovsky D,Le JL,et al.Expression of the coxsackie adenovirus receptorin normal prostate and in primary and metastatic prostate carcinoma:potentialrelevance to gene therapy[J].Cancer Res,2002,2:3812.
[12]Kitazono M,Goldsmith M E,Aikou T,et al.Enhancedadenovirus transgene in malignant cells treated with the histone deacetylaseinhibitorFR901228[J].CancerRes,2001,61:6328.
