固定化重组细胞催化非天然二肽合成
2010-06-04程仕伟,魏东芝,林金萍
近年来二肽已成为药物、保健食品、食品添加剂工业的研发新热点,应用前景广阔。例如二肽经甲酯化和环化后生成哌嗪二酮衍生物,多种哌嗪二酮类化合物对DNA拓扑异构酶Ⅱ有特异性抑制作用[1],可筛选为潜在的抗肿瘤药物。目前,二肽合成多采用化学法,但是化学法合成二肽要求有保护和活化步骤,反应步骤长、副产物多[2]。利用酶作为催化剂合成二肽的工艺相对简单,产品质量高,对环境友好,因而受到广泛重视且部分品种已实现产业化。如荷兰DSM公司和日本Tova Soda公司利用蛋白酶大规模合成增甜剂阿斯巴甜,目前年生产能力在1万 t左右[3]。虽然蛋白酶被成功用于二肽合成,但其只能选择性合成L-氨基酸衍生物,而不能转化非天然氨基酸如D-苯甘氨酸[4]。
青霉素酰化酶是半合成抗生素生产中的关键酶,可以合成众多的β-内酰胺抗生素[5,6]。此外,该酶在手性合成物拆分与合成方面也应用广泛[7,8]。Khimiuk等[2]发现E.coli来源的青霉素G酰化酶(Penicillin G acylase,PGA) 可用于酶法合成非天然的苯甘氨酸二肽,其合成过程如图1所示[2]。
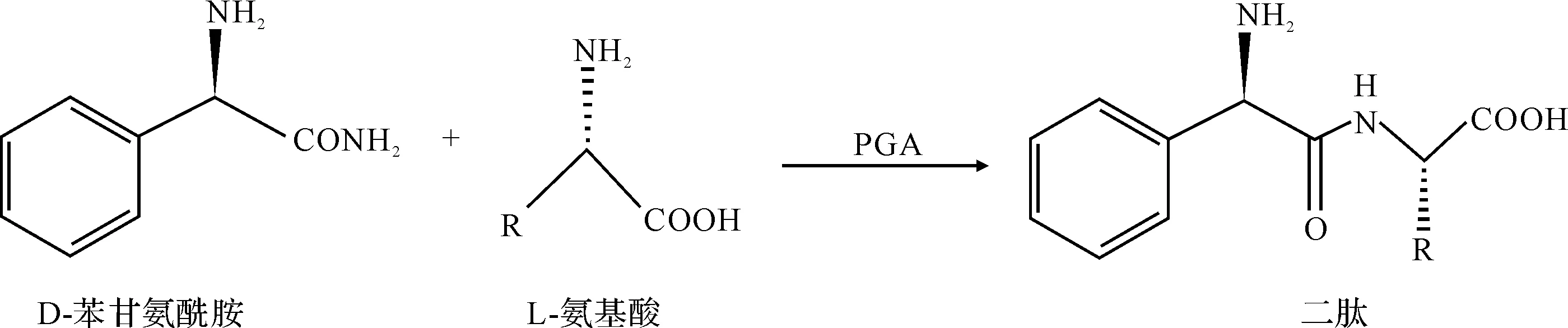
图1 青霉素G酰化酶催化合成二肽
由于A.faecalis来源的 PGA在底物选择性、热稳定性、有机溶剂耐受性等方面具有独特优势[9,10],催化性能优于E.coliPGA,因此,作者采用A.faecalisPGA的固定化重组细胞作为催化剂合成D-苯甘氨酸二肽,拟为其规模化酶法生产打下良好基础。
1 实验
1.1 菌株、试剂和仪器
产A.faecalisPGA的重组E.coli菌株由中国科学院袁中一研究员提供。
L-亮氨酸、L-异亮氨酸,生物纯,上海求德生物化工有限公司;L-组氨酸、L-丙氨酸、L-谷氨酸、L-甘氨酸,生物纯,上海化学试剂有限公司;D-苯甘氨酰胺(纯度>99%),浙江天台药业。
1100 Series型高效液相色谱仪,Agilent 公司;722S型可见分光光度计,上海菁华;HZ-8212S型恒温振荡水浴,华利达;自动控温控pH值的酶催化反应装置,自制。
1.2 方法
1.2.1 菌体细胞通透处理
离心收集细胞,在磷酸缓冲溶液(50 mmol·L-1,pH值7.8)中重悬浮。加入一定量的溴化十六烷基三甲基氯化铵(CTAB),在搅拌条件下处理细胞。离心,去除上清液,得渗透处理的细胞。
1.2.2 重组细胞固定化
取处理的细胞悬浮于Tris-HCl缓冲溶液中(50 mmol·L-1,pH值8.0),在37℃下依次加入10 mL 5% 海藻酸钠及10 mL 15%聚乙烯醇,混合。将混匀物逐滴滴入含5% CaCl2的冷饱和硼酸溶液中,在4℃静置72 h。抽滤去除硼酸溶液,用Tris-HCl缓冲溶液清洗几次。
包埋细胞和终浓度2%(体积分数)的戊二醛在4℃ 下交联反应6 h,然后用Tris-HCl缓冲溶液(50 mmol·L-1,pH值8.0)清洗几次,即得固定化细胞。
1.2.3 生物催化合成二肽
取50 mmol·L-1的D-苯甘氨酰胺和65 mmol·L-1的L-氨基酸,调pH值9.5,定容至20 mL,加入5 g固定化细胞进行反应,控制反应温度为25℃、pH值为9.5。在反应不同阶段定时取样,参照Khimiuk等[2]方法用HPLC监测反应进程及各物质的含量。反应完毕,过滤去除固定化细胞,加入2 mol·L-1的H2SO4调pH值至5.0。静置12 h,二肽逐渐沉淀析出,过滤,得目的产物。
反应液通过装有XDB-C18柱的Agilent 1100系列HPLC液相色谱系统检测。流动相:含40%(体积分数) 乙腈、0.7 g·L-1SDS的磷酸溶液(pH值3.5),流速1.0 mL· min-1,柱温 20℃,检测波长208 nm。
1.2.4 酶活测定
酶活的测定采用PDAB法[11]:称取一定量的固定化细胞,在37℃下预热5 min,加入预热的4%青霉素G钾盐溶液5 mL。反应5 min后立即取0.5 mL于空白试管中,加入3.5 mL PDAB显色液显色5 min,在415 nm下比色测定。
2 结果与讨论
2.1 细胞通透处理及固定化载体选择
由于青霉素G酰化酶在大肠杆菌中主要位于细胞周质内[12],要提高固定化细胞的酶活性需先进行重组细胞通透处理,以减少细胞对酶的扩散限制。CATB可作用于大肠杆菌细胞的脂质和磷脂层[13],导致细胞蛋白结构发生变化,进而增加细胞的通透性。通过优化CTAB处理条件,在0.3%(质量体积比)CTAB、25℃、处理75 min的条件下,细胞通透性增加了7.5倍。
不用戊二醛交联的聚乙烯醇固定化细胞酶活虽较高[21.8 U·(g湿载体)-1],但在碱性反应条件下稳定性较差,批反应次数较少。采用戊二醛交联聚乙烯醇固定化细胞虽酶活降低约1/3,但是固定化细胞稳定性好、硬度高,形成多空颗粒,且细胞大小基本相同,呈规则的球形。在pH值10、50℃下催化青霉素G钾盐的重复利用实验中,每150 min为一批次,使用15批次基本没有酶失活,重复31批次后仍保留65%的酶活,37批次后仍有较好的弹性和较高的硬度[14]。此外,还尝试了采用海藻酸钠固定法及海藻酸钠-明胶交联法固定细胞,获得的催化剂在硬度、稳定性等方面均相对较差。
2.2 HPLC检测方法的建立
产物形成过程均采用HPLC方法监测,大多可形成4个有规律的峰。以D-Phenylglyl-L-isoleucine为例,其HPLC图谱见图2。
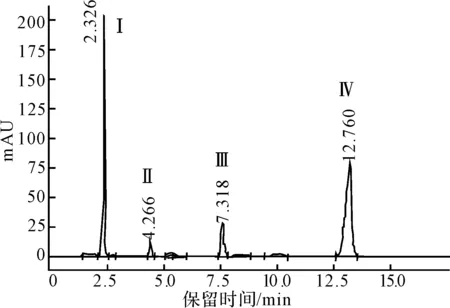
Ⅰ.D-Phenylglycine Ⅱ.L-Isoleucine Ⅲ.D-Phenylglycine amide Ⅳ.D-Phenylglyl-L-isoleucine
反应过程中各物质的保留时间见表1。
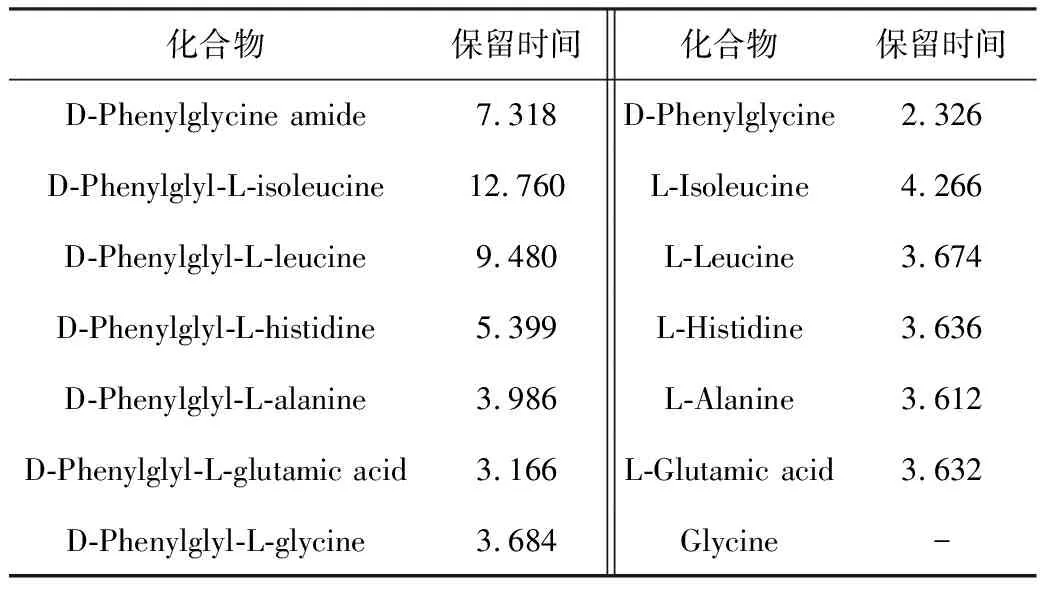
表1 HPLC分析中各物质的保留时间/min
2.3 二肽合成反应过程解析
由于D-苯甘氨酰胺本身就是青霉素G酰化酶的水解底物,所以反应系统中除了D-苯甘氨酰胺与L-氨基酸生成二肽的合成反应外,还存在D-苯甘氨酰胺自身被酶水解生成D-苯甘氨酸和氨气的反应以及合成的二肽被酶水解的反应,故整个反应中D-苯甘氨酸为主要反应副产物,应尽量避免该副产物的形成。在此,为减少D-苯甘氨酰胺的水解,采用L-氨基酸底物过量的方法。
以D-苯甘氨酰胺为固定底物,利用固定化A.faecalisPGA重组细胞为催化剂,选取不同L-氨基酸进行二肽合成实验,其中几种效果较好二肽的合成过程见图3。
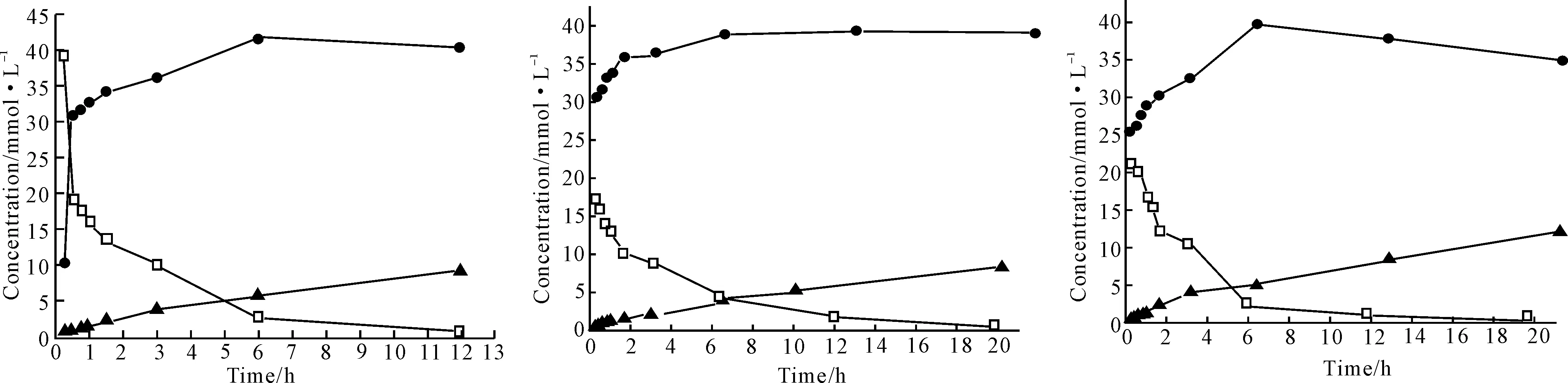
a.D-Phenylglyl-L-glycine b.D-Phenylglyl-L-isoleucine c.D-Phenylglyl-L-alanine

d.D-Phenylglyl-L-glutamic acid e.D-Phenylglyl-L-histidine f.D-Phenylglyl-L-leucine
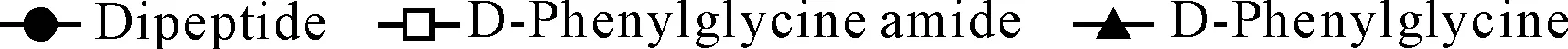
图3 固定化重组细胞催化合成二肽的过程图
由图3可以看出,采用固定化A.faecalisPGA重组细胞催化合成二肽时,反应速度较快。在相同反应条件下,反应15 min时,除D-Phenylglyl-L-glycine的浓度为10.39 mmol·L-1(液相得率20%)外,其余5种二肽的浓度均超过25 mmol·L-1,即液相得率超过了50%。
整个反应过程中,主要副产物D-苯甘氨酸生成的过程曲线大致相似,开始时由于参加二肽合成的L-氨基酸浓度较高,酶主要催化二肽合成反应,故副产物D-苯甘氨酸的生成量较少;随着反应时间的延长,游离的L-氨基酸转化为相应二肽而浓度变小,进而D-苯甘氨酰胺的水解反应速度加快,副产物D-苯甘氨酸浓度逐渐加大。因此,在选取固定底物D-苯甘氨酰胺与游离L-氨基酸的摩尔比时,加大游离L-氨基酸摩尔量 [理论上n(D-苯甘氨酰胺)∶n(氨基酸)=1∶1.5]可使D-苯甘氨酰胺尽可能多地参加二肽合成反应,在一定程度上减少副产物D-苯甘氨酸的生成。
由图3还可以看出,二肽合成与副产物D-苯甘氨酸生成存在一定的关系,6种非天然二肽获得最大生成量的时间均在反应6 h左右。当达到最大产物量后,产率稳定持续一段时间或开始逐渐下降。相对应的,副产物D-苯甘氨酸的生成量开始逐渐加大。当二肽合成获得最大得率时,副产物D-苯甘氨酸的得率均为5 mmol·L-1左右,相当于10%的D-苯甘氨酰胺参加了水解反应。
Khimiuk等[2]认为在副产物D-苯甘氨酸浓度10%左右终止反应较为合适。本实验发现,D-苯甘氨酸浓度在10%左右时,虽可获得最大二肽得率,但反应时间较长(6 h)。鉴于二肽合成反应初期速率较快、副产物D-苯甘氨酸较少,从经济角度考虑,作者认为终止反应的时间在60 min左右较为合适,此时D-苯甘氨酸量少,易与目的产物分离。
青霉素G酰化酶的酰基连接位点对苯乙酸及其衍生物有高度选择性,也可以接受苯甘氨酸派生物作为酰基供体。相反地,其亲核连接端对大部分氨基酸的L-异构体具有广泛的选择性,因此,青霉素G酰化酶在合成D-苯甘氨酸二肽方面具有独特的优势[2,3]。
2.4 二肽合成的结果比较
二肽合成的最高液相得率、实际产品分离率、反应后残余酶活(20 h)比较见表2。
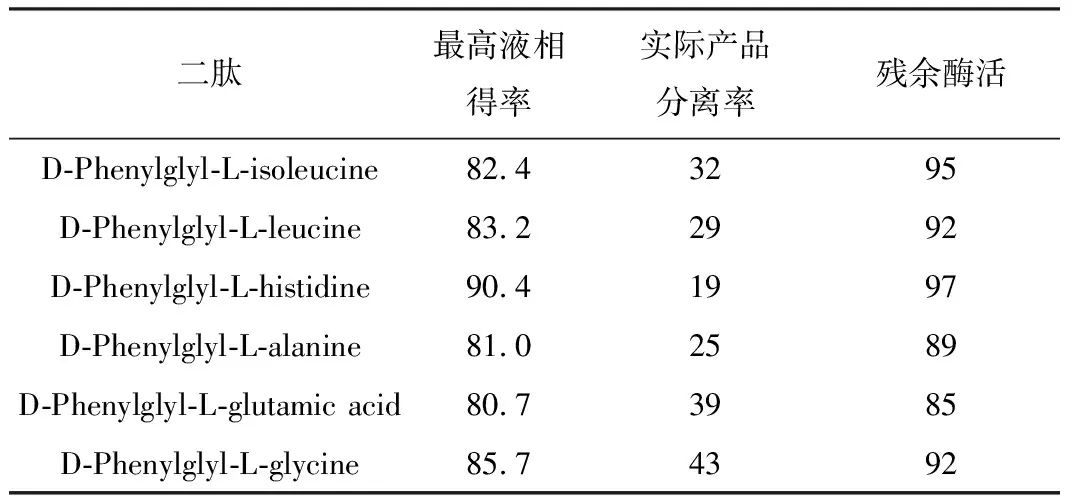
表2 二肽合成的结果比较/%
由表2可以看出,6种二肽合成过程获得的最高液相得率较高,均超过80%,D-Phenylglyl-L-histidine的液相得率甚至超过90%。将反应溶液的pH值降至5.5左右时,虽然产物逐渐析出,但是过滤干燥后称量发现其实际的产品分离率均不高,其中最高的D-Phenylglyl-L-glycine二肽的实际产品分离率仅43%,近一半的二肽仍然在反应溶液中没有析出。D-Phenylglyl-L-histidine二肽的实际产品分离率仅为19%,还有大约72%的产品没有析出。
综上所述,虽然固定化A.faecalisPGA重组细胞催化合成二肽具有反应速度快、液相得率较高的优势,但是产品的实际分离率相对较低,产品的分离方法还有待深入研究。
由表2还可以看出,反应完毕后酶活丢失较少,酶活丢失最多的D-Phenylglyl-L-glutamic acid二肽合成反应,残余酶活为85%,表明固定化细胞可重复使用。Shcherbakova等[15]发现E.coliPGA在高苯甘氨酰胺浓度下严重失活,苯甘氨酰胺浓度为100 mmol·L-1、反应100 min时,残余酶活仅为25%左右。而以A.faecalisPGA为催化剂,同样反应条件下的残余酶活在62%左右,表明A.faecalisPGA在二肽合成方面比E.coliPGA具有更高的底物稳定性,应用潜力较好。
3 结论
选取具有代表性的6种L-氨基酸作为酰基受体,以D-苯甘氨酰胺作为酰基供体,采用固定化A.faecalisPGA重组细胞进行催化二肽合成的研究。结果表明,催化反应的速度较快,除D-Phenylglyl-L-glycine二肽开始时反应速率稍慢(15 min时液相得率20%) 外,其余几种二肽的合成在反应15 min时液相得率均超过50%。合成的二肽基本在反应6 h时得到最高液相产率,均超过80%。虽然反应过程获得的液相得率较高,但实际产品分离率并不高,关于产品的分离纯化方法还有待继续进行研究。
参考文献:
[1] Hasinoff B B,Kuschak T I,Yalowich J C,et al.A QSAR study comparing the cytotoxicity and DNA topoisomerase Ⅱ inhibitory effects of bisdioxopiperazine analogs of ICRF-187(dexrazoxane)[J].Biochem Pharmacology,1995,50(7):953-958.
[2] Khimiuk A Y,Korennykh A V,van Langen L M,et al.Penicillin acylase-catalyzed peptide synthesis in aqueous medium:A chemo-enzyme route to steroisomerically pure diketopiperazines[J].Tetrahedron:Asymmetry,2003,14(20):3123-3128.
[3] Liese A,Seelbach K,Wandrey C.Industrial Biotransformations (2nd)[M].Wiley-VCH,2006:320-322.
[5] Wei D Z,Liu Y.Effects of ethylene glycol on the synthesis of ampicillin using immobilized penicillin G acylase[J].J Chem Technol Biot,2003,78(4):431-436.
[6] Liu Y,Wei D Z,Zhang Y W.Semi-continuous enzymatic synthesis of cefaclor enhanced by in situ product removal[J].J Chem Technol Biot,2004,79(5):480-485.
[7] Guranda D T,Khimiuk A K,van Langen L M,et al.An′easy-on,easy-off′protecting group for the enzymatic resolution of(±)-1-phenylethylamine in an aqueous medium[J].Tetrahedron :Asymmetry,2004,15(18):2901-2906.
[8] Rocchietti S,Urrutia A S V,Pregnolato M,et al.Influence of the enzyme derivative preparation and substrate structure on the enantioselectivity of penicillin G acylase[J].Enzyme Microb Technol,2002,31(1-2):88-93.
[9] Verhaeart R M D,Riemins A M.Molecular cloning and analysis of the gene encoding the thermostable penicillin G acylase fromAlacaligenesfaecalis[J].Appl Environ Microb,1997,63(9):3412-3418.
[10] van Langen L M,Oosthoek N H P,Guranda D T,et al.Penicillin acylase-catalyzed resolution of amines in aqueous organic solvents[J].Tetrahedron:Asymmetry,2000,11(22):4593-4600.
[11] Shewale J G,Kumar K K,Ambekar G R.Evaluation of determination of 6-amino penicillanic acid byp-dimethylaminobenzadehyde[J].Biotechnol Tech,1987,1(1):69-72.
[12] Chou C P,Yu C C,Tseng J H,et al.Genetic manipulation to identify limiting steps and develop strategies for high-level expression of penicillin acylase inEscherichiacoli[J].Biotechnol Bioeng,1999,63(3):263-272.
[13] Mutalik R B,Gaikar V G.Cell permeabilization for extraction of penicillin acylase fromEscherichiacoliby reverse micellar solutions[J].Enzyme Microb Technol,2003,32(1):14-26.
[14] Cheng S W,Wei D Z,Song Q X,et al.Immobilization of permeabilized whole cell penicillin G acylase fromAlcaligenesfaecalisusing pore matrix crosslinked with glutaraldehyde[J].Biotechnol Lett,2006,28(14):1129-1133.
[15] Shcherbakova T A,Korennykh A V,van Langen L M,et al.Use of high acyl donor concentrations leads to penicillin acylase inactivation in the course of peptide synthesis[J].J Mol Catal B-Enzym,2004,31(1-3):63-65.
