钒氧阴离子团簇与小分子碳氢化合物反应的实验和理论研究
2010-03-06马嘉璧吴晓楠赵艳霞何圣贵丁迅雷
马嘉璧 吴晓楠 赵艳霞 何圣贵 丁迅雷
(1中国科学院化学研究所,分子动态与稳态结构国家重点实验室,北京 100190;2中国科学院研究生院,北京 100049)
Vanadium oxide based catalysts have been widely used in the oxidation of hydrocarbon molecules[1-2].To understand the mechanisms of these reactions on the active sites of the catalytic surface,great efforts have been devoted to investigating the reactivity of vanadium oxide clusters toward various hydrocarbon molecules in gas phase using mass spectrometry based experimental techniquesin conjunction with theoreticalcalculations[3-17].
Most of the studies were focused on cationic vanadium oxide clusters[3-8,15-17].It has been found that stoichiometric(n= 1-5)clusters are able to not only transfer one oxygen atom to unsaturated hydrocarbon molecules[5-8,17],but also abstract one hydrogen atom from methane[15-16,18].The reactivity of neutral vanadium oxide clusters toward hydrocarbons has been studied based on single photon ionization in conjunction with theoretical calculations,recently[9-13].Oxygen rich vanadium oxide clusters VO3(V2O5)n(n=0-2)exhibit particular reactivity with regard to the C=C bond cleavage of alkenes.It has been demonstrated that anionic clusters are much less reactive than the corresponding cationic ones[6,19],and only very limited works have been reported.Castleman and co-workers[6]found that no association or oxygen transfer products were observed in the mass spectra when vanadium oxide cluster anions reacted with ethane and ethylene,while in reactions with butene or butadiene(V2O5)nO-(n=1,2)and V3O-9displayed a minor oxygen transfer reaction channel[3].Li and coworkers[14]identified oxygen transfer product C3H6O from reaction of V4O-11with propylene under the condition of photon radiation.Further theoretical work studied the reaction mechanisms and suggested that the product C3H6O was mainly propylene oxide[20].
To have a good understanding of the chemistry of O●species over vanadium as well as other metal oxide clusters,it is important to devote more efforts to the much less studied anionic cluster systems.In this work,reactions of vanadium oxide cluster anions with ethane(C2H6),n-butane(C4H10),ethylene(C2H4),and propylene(C3H6)were studied by experiments based on time of flight(TOF)mass spectrometry.The Δ=1 anions(V2O5)nO-(n=1,2)are found to be able to abstract H atom from alkanes while produce association products with alkenes.Further theoretical calculations using the density functional theory(DFT) were performed to study the reaction mechanisms.
1 Methods
1.1 Experimental methods
The experimental setup for a pulsed laser ablation/supersonic nozzle coupled with a fast flow reactor is similar to the one described in previous studies[22-23].Only a brief description of the experiment is given here.Theclusters were generated by laser ablation of a rotating and translating vanadium metal disk (99.5%purity)in the presence of about 1%O2seeded in a He carrier gas with backing pressure of 300 kPa.A 532 nm(second harmonic of Nd3+:yttrium aluminum garnet-YAG)laser with energy of 5-8 mJ per pulse and repetition rate of 10 Hz was used. The gas was controlled by a pulsed valve(General Valve,Series 9).Possibly due to residual water in gas handling system,undesirable hydroxo species,such as(z>0 and M is a metal atom)often occur in the distribution of transition metal oxide clusters[24]although high purity gases(He and O2,99.995%)are used.To eliminate or to significantly decrease contributions of these undesirable species in our cluster distributions,the prepared gas mixture(O2/He)was passed through a 10 m long copper tube foil at low temperature(T=77 K)before entering into the pulsed valve.By using the method in Ref.[25],the instantaneous total gas pressure in the fast flow reactor was estimated to be around 3×102Pa at T=350 K.The cluster vibrational temperature is believed to be close to the ambient temperature that is around 300-400 K considering that the carrier gas can be heated during the process of laser ablation[26].The clusters formed in a gas channel(2 mm(diameter)×25 mm(length))were expanded and reacted with hydrocarbon molecules(C2H6,C4H10,C2H4, and C4H8,99.9%)in a fast flow reactor(6 mm(diameter)×60 mm (length)).The reactant gas was pulsed into the reactor by a second pulse valve.After reacting in the fast flow reactor,the reactant and product ions exiting from the reactor were skimmed(3 mm(diameter))into a vacuum system of a TOF mass spectrometer for mass(to charge ratio)measurement.Ion signals were generated by a dual micro-channel plate detector and recorded with a digital oscilloscope(LeCroy WaveSurfer 62Xs)by averaging 500-1000 traces of independent mass spectra(each corresponds to one laser shot).
1.2 Computational methods
The DFT calculations using the Gaussian 03 program[27]were employed to study the reaction mechanisms ofandThe hybrid B3LYP exchange-correlation function-al[28-29]was adopted.A set of contracted Gaussian basis set of triple zeta valence quality[30]plus one p function was used for H and V atoms and one d function for C and O atoms(denoted as TZVP in Gaussian 03).Geometry optimization with full relaxation of all atoms was performed.Transition state optimization was performed by using the Berny algorithm[31]from an initial structure obtained through a potential energy surface scan with an appropriate internal coordinate.Vibrational frequency calculations were performed to check that reaction intermediates and transition state species have zero and only one imaginary frequency,respectively.Intrinsic reaction coordinate calculations[32-33]were also performed to confirm that each transition state connected two appropriate local minima in the reaction paths.The zero-point vibration corrected energies(ΔE0K)and the Gibbs free energies at 298 K(ΔG298K)were reported in this work. Cartesian coordinates,electronic energies,and vibrational frequencies for all of the optimized structures are available upon request.
2 Results
2.1 Experimental results
The TOF mass spectra for the reactions ofclusters with C2H6and C4H10are plotted in Fig.1.The left panel(Fig.1(a,b,c)) shows the results in theAfter the reaction with C2H6(Fig.1(b)),a new mass peak that can be assigned as V2O6H-is observed.Similar results are obtained for the reactions with C4H10(Fig.1(c)).The peak intensity of V2O6H-relative toin the reaction with C4H10is more intense than that in the reaction with C2H6.Spectra in theregion are plotted in the right panel(Fig.1(d,e,f)).The mass resolution in this region is not as good as that in the,but the appearance of a product peak that can be assigned as V4O11H-is apparent.

Fig.1 TOF mass spectra in two selected mass regions for reactions of VxO-ywith He(a and d),55 Pa C2H6(b and e), and 42 Pa C4H10(c and f)in the fast flow reactorNumbers x,y denote VxO-yand x,yX denote VxOyX-in which X=H or H2O.
Except for V2O6H-and V4O11H-,no association products(such asandor other reaction products can be found in the experimental spectra.The previous theoretical study indicates thatand(Δ=1 cluster series)are oxygen centered radicals[15]which can be very oxidative in the reaction with hydrocarbon molecules.Since the saturated hydrocarbons C2H6and C4H10are quite stable molecules,the observation of hydrogen abstraction products V2O6H-and V4O11H-by the experiments(Fig.1)suggests thatand Vare the reactant clusters,respectively.
The TOF mass spectra for the reactions of VxO-yclusters with C2H4and C3H6are plotted in Fig.2.Association products are found only forandwith both C2H4(Fig. 2(b))and C3H6(Fig.2(c)).No apparent appearance of hydrogen abstraction products,such as V2O6H-and V4O11H-in Fig.1,is observed.
2.2 Computational results
The ground state structures ofandcan be found in literature[15,34-35].The reaction path of+C4H10is calculated and the results are plotted in Fig.3.The initial interaction betweenand C4H10involves a weak hydrogen bonding(binding energy,Eb=0.08 eV)with which the reaction intermediate 1 (denoted as I1)forms.Thecluster has Cssymmetry and the spin densities are located at two terminal oxygen atoms denoted as O1 and O2 in Fig.3.The bond lengths of V—O1 and V—O2 are 0.172 and 0.167 nm,respectively.It is expected that V—O1 is more reactive than V—O2 since the former is longer than the latter and there are more spin densities on O1 than on O2.However,Fig.4 shows that the intra-cluster spin density re-distribution in,which results in lengthening of V—O2 and shortening of V—O1,is very easy to take place.As a result,both V—O1 and V—O2 can be the active sites of the cluster.Very careful tests were carried out to optimize an alternative I1 structure,in which the length of the V—O bond that interacts with the hydrogen atom(such as H1 of I1 in Fig.3)is between 0.168 and 0.178 nm.All of such tests failed and the DFT optimizations ended with the I1 structure.This is probably because the terminal O atom with shorter V—O distance has more negative charge,which favors the formation of the hydrogen bonding.

Fig.2 TOF mass spectra for reactions of V2-4O-ywith He(a), C2H4(b),and C3H6(c)Numbers x,y denote VxO-yand x,yX denote VxOyX-in which X=C2H4or C3H6.

Fig.3 Energy diagram for the reaction of V2O-6with C4H10The values are relative to the entrance channel.The data in the parentheses refer to ΔE0Kand ΔG298Kin eV,respectively.Some of the bond lengths in nm are given.
After formation of I1,one H atom can be abstracted from a methylene group of C4H10by surmounting a small overall reaction barrier(ΔE0K=0.01 eV for TS12),which leads to formation ofa weaklybonded speciesI2(V2O6H…C4H9)-.The desorption of C4H9from V2O6H-needs only 0.16 eV energy.The H atom abstraction from the methyl group of C4H10is also possible and the calculated overall barrier is 0.08 eV(ΔE0Kvalue),which is slightly higher than the energy of TS12 in Fig.3.Note that hydrogen abstraction from the methyl group to formV2O6H-+1-C4H9,ΔH0K=-0.32 eV)is also thermodynamically less favorable than that the abstraction from the methylene group to form 2-C4H9(ΔH0K=-0.50 eV).Similar calculations were performed for reactionThe overall barrier for the H atom abstraction is 0.10 eV,which is very close to the barrier of methyl group activation in C4H10.
For reactions of vanadium oxide cluster anions with alkenes, the reaction mechanisms for the simplest systemwere studied and the results are given in Fig.5.At first,the C2H4molecule weakly adsorbed on V2O6-through two hydrogen bonds (I3).A small overall barrier of 0.17 eV(TS34)has to be overcome to activate C=C double bond and form species I4 with a C—O bond(note that the distance between the two C atoms is increased significantly from 0.135 nm(in I3)to 0.149 nm(in I4)). Then a rotation around the C—O bond in I4 takes place and the distance between the other pair of C and O atoms is shorten to make a second C—O bond.A five-membered ring(-V-O-C-CO-)structure with binding energy of 1.64 eV is formed as the association product in the reaction ofwith C2H4.Because TS45 is significantly lower in energy than TS34,the rate determining step of the whole reaction is the activation of C=C double bond or the formation of the first C—O bond.Note that the DFT calculations indicate that the enthalpy change(ΔH0K)forV2O6H-+C2H3is 0.09 eV,indicating that hydrogen atom abstraction from C2H4is thermodynamically impossible,in qualitative agreement with the non-observation of V2O6H-in the reaction with C2H4(Fig.2).
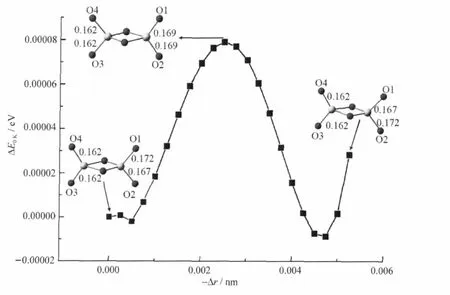
Fig.4 Relaxed potential energy curve by fixing the bond length of V—O1 in V2O-6at different values and optimizing all of the other degrees of freedomΔr is the relative change of V—O1 bond length.

Fig.5 Energy diagram for the reaction of V2O-6with C2H4 See the caption of Fig.3 for explanations.
3 Discussion
The anionic clusters were considered to be inert toward hydrocarbon molecules because the presence of negative charge may reduce electron withdrawing effects and decrease the polarizations on small molecules[6].However,experiments and DFT calculations suggest that the anionic clustersandcan also react with small hydrocarbon molecules by abstracting one hydrogen atom from alkanes C2H6and C4H10and producing association products with alkenes C2H4and C3H6.
It should be pointed out that the rate constants of anionic clustersand[or(V2O5)1,2O-]with the studied molecules are much less than those of cationic clusterswith small molecules such as CH4.Fig.1(c)shows that 42 Pa C4H10should be used to generated product V2O6H-with intensity being comparable to the reactant clusterwhile the previous study[16]using the same apparatus for+CH4indicated that much less pressure(0.25 Pa)of CH4should be used to generatecouple with comparable intensities.
The number of collisions that thecluster(effective diameter≈0.64 nm)experiences with 42 Pa C4H10(effective diameter≈0.66 nm)in the fast flow reactor(length=60 mm)is about 692,which causes significant scattering of un-reacted(and also the product V2O6H-)from the cluster beam.Further increase of the reactant(C2H6and C4H10)gas pressures in the reactor results in relative signal increase of V2O6H-versuswhile the absolute signal of V2O6H-becomes very weak.Deuterated n-butance(C4D10)was tested to generate product that can be assigned as V2O6D-(m/z=200)and V4O11D-(m/z=382).Because of kinetic isotopic effect(slower activation of C—D versus C—H)and the scattering of the clusters and reaction products by C4D10,the intensities of the peaks at 200 and 382(m/z)are very weak.To discard the possibility that V2O6H-is the product from the cluster reactions with trace amount of H2O in reactant gas,H2O(by replacing C2H6or C4H10)was reacted with VxO-yin the fast flow reactor.In this case,we did see weak signal of V2O6H-while very strong signal of V2O6H-2(V2O5H2O-)also appeared.In contrast, V2O6H-2is much weaker than V2O6H-in Fig.1(b,c),indicating H2O is not the hydrogen source to produce V2O6H-.
By using the method in the previous studies[16,25,36]and assuming that deplection ofand V2O6H-by the C4H10is the same, the first order rate constant(k1)for+C4H10→V2O6H-+2-C4H9is estimated to be about 1×10-12cm3·molecule-1·s-1,which is smaller than k1for+CH4→V4O10H++CH3(1.3×10-10[16]or 5.6× 10-10cm3·molecule-1·s-1[18])by two to three orders of magnitude.
The above relative rate constants for anionic and cationic reaction systems are consistent with the DFT results of the reaction mechanisms.Fig.3 shows that hydrogen atom abstraction from C4H10byis subject to small overall reaction barrier while the DFT study in Ref.[18]predicted that hydrogen atom abstraction from CH4byis barrierless and no transition state(such as TS12 in Fig.3)for CH activation can be located. Note that a significant overall free energy barrier(TS12,ΔG298K= 0.50 eV)is predicted forGiven that the reaction intermediate I1 is not fully at thermal equilibrium due to the relatively low pressure(300 Pa)in the reaction cell,and considering the fact that the initially formed meta-stable species(I1)*carries center of mass collisional energy(Ek=0.23 eV forapproaching C4H10with velocity of 1 km·s-1)and cluster vibrational energy(Evib=0.23 eV by DFT forat 350 K),it is still possible to observe the hydrogen atom abstraction reaction+ C4H10→V2O6H-+C4H9although the rate is very slow relative to
Fig.2 indicates that the reactions between(or V)and alkenes C2H4and C3H6are ended with the association products through 3+2 cycloaddition reaction.No further reaction products,such as(of which the relative signal does not increase upon the reaction)for the oxygen transfer reaction,for the C=C bond cleavage reaction,are found.This is different from the reactivity of cationic or neutral vanadium oxide clusters[5-14,16].To verify our experimental findings,theoretical calculations are performed to check the possibilities for the following reactions:
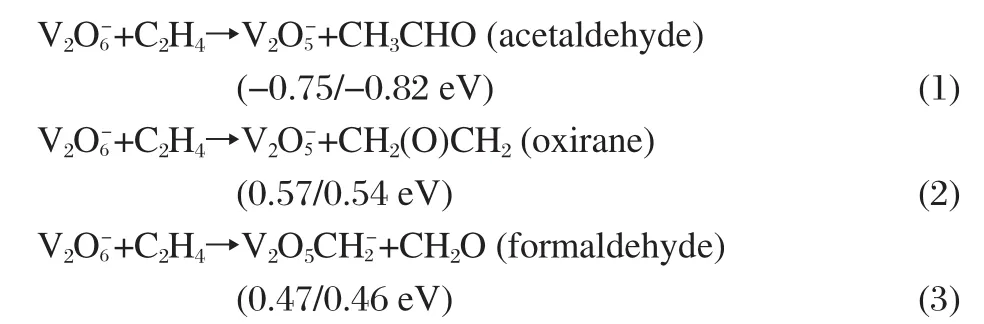
The changes of the zero-point vibration corrected energies and the Gibbs free energies of these reactions(ΔH0K/ΔG298K)are listed after each equation.Reaction(1)is an oxygen transfer reaction and thermodynamically permitted.This reaction involves a hydrogen transfer from one C atom to the other in C2H4.We have studied the hydrogen transfer process from both I4 and P in Fig.5,and the relative energies(ΔH0K/ΔG298K)of the transition states involved with the two initial structures are(0.62/1.08 eV) and(0.60/1.06 eV),respectively.So reaction(1)is kinetically forbidden.Reaction(2)is also an oxygen transfer reaction and reaction(3)is a C=C bond cleavage reaction,both of which are thermodynamically forbidden.Thus,the theoretical calculations further suggest that in the experiments only the association productcan be found for
4 Conclusions
1 Weckhuysen,B.M.;Keller,D.E.Catal.Today,2003,78:25
2 Centi,G.;Trifiro,F.;Ebner,J.R.;Franchetti,V.M.Chem.Rev., 1988,88:55
3 Bell,R.C.;Castleman Jr.,A.W.J.Phys.Chem.A,2002,106: 9893
4 Feyel,S.;Schroder,D.;Schwarz,H.J.Phys.Chem.A,2006,110: 2647
5 Justes,D.R.;Mitric,R.;Moore,N.A.;Bonacic-Koutecky,V.; Castleman Jr.,A.W.J.Am.Chem.Soc.,2003,125:6289
6 Zemski,K.A.;Justes,D.R.;Castleman Jr.,A.W.J.Phys.Chem. A,2001,105:10237
7 Moore,N.A.;Mitric,R.;Justes,D.R.;Bonacic-Koutecky,V.; Castleman,A.W.J.Phys.Chem.B,2006,110:3015
8 Justes,D.R.;Castleman,A.W.;Mitric,R.;Bonacic-Koutecky,V. Eur.Phys.J.D,2003,24:331
9 Ma,Y.P.;Xue,W.;Wang,Z.C.;Ge,M.F.;He,S.G.J.Phys. Chem.A,2008,112:3731
10 Dong,F.;Heinbuch,S.;Xie,Y.;Bernstein,E.R.;Rocca,J.J.; Wang,Z.C.;Ding,X.L.;He,S.G.J.Am.Chem.Soc.,2009,131: 1057
11 Dong,F.;Heinbuch,S.;Xie,Y.;Rocca,J.J.;Bernstein,E.R.; Wang,Z.C.;Deng,K.;He,S.G.J.Am.Chem.Soc.,2008,130: 1932
12 Wang,Z.C.;Xue,W.;Ma,Y.P.;Ding,X.L.;He,S.G.;Dong,F.; Heinbuch,S.;Rocca,J.J.;Bernstein,E.R.J.Phys.Chem.A,2008, 112:5984
13 Ma,Y.P.;Ding,X.L.;Zhao,Y.X.;He,S.G.ChemPhysChem, 2010,11:1718
14 Li,S.H.;Mirabal,A.;Demuth,J.;Woste,L.;Siebert,T.J.Am. Chem.Soc.,2008,130:16832
15 Zhao,Y.X.;Ding,X.L.;Ma,Y.P.;Wang,Z.C.;He,S.G.Theor. Chem.Acc.,2010,DOI:10.1007/s00214-010-0732-8
16 Zhao,Y.X.;Wu,X.N.;Wang,Z.C.;He,S.G.;Ding,X.L.Chem. Commun.,2010,46:1736
17 Yin,S.;Ma,Y.P.;Du,L.;He,S.G.;Ge,M.F.Chin.Sci.Bull., 2008,53:3829
18 Feyel,S.;Dobler,J.;Schroder,D.;Sauer,J.;Schwarz,H.Angew. Chem.Int.Edit.,2006,45:4681
19 Johnson,G.E.;Mitric,R.;Nossler,M.;Tyo,E.C.;Bonacic-Koutecky,V.;Castleman,A.W.J.Am.Chem.Soc.,2009,131: 5460
20 Li,H.B.;Tian,S.X.;Yang,J.L.Chem.Eur.J.,2009,15:10747
21 Lunsford,J.H.Catal.Rev.Sci.Eng.,1973,8:135
22 Xue,W.;Yin,S.;Ding,X.L.;He,S.G.;Ge,M.F.J.Phys.Chem. A,2009,113:5302
23 Yin,S.;Xue,W.;Ding,X.L.;Wang,W.G.;He,S.G.;Ge,M.F. Int.J.Mass Spectrom.,2009,281:72
24 Johnson,G.E.;Mitric,R.;Tyo,E.C.;Bonacic-Koutecky,V.; Castleman,A.W.J.Am.Chem.Soc.,2008,130:13912
25 Xue,W.;Wang,Z.C.;He,S.G.;Xie,Y.;Bernstein,E.R.J.Am. Chem.Soc.,2008,130:15879
26 Geusic,M.E.;Morse,M.D.;Obrien,S.C.;Smalley,R.E.Rev.Sci. Instrum.,1985,56:2123
27 Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03. Revision B.05.Wallingford,CT:Gaussian Inc.,2003
28 Becke,A.D.J.Chem.Phys.,1993,98:5648
29 Lee,C.T.;Yang,W.T.;Parr,R.G.Phys.Rev.B,1988,37:785
30 Schafer,A.;Huber,C.;Ahlrichs,R.J.Chem.Phys.,1994,100: 5829
31 Schlegel,H.B.J.Comput.Chem.,1982,3:214
32 Gonzalez,C.;Schlegel,H.B.J.Chem.Phys.,1989,90:2154
33 Gonzalez,C.;Schlegel,H.B.J.Phys.Chem.,1990,94:5523
34 Santambrogio,G.;Brummer,M.;Woste,L.;Dobler,J.;Sierka,M.; Sauer,J.;Meijer,G.;Asmis,K.R.Phys.Chem.Chem.Phys.,2008, 10:3992
35 Gong,Y.;Zhou,M.F.;Andrews,L.Chem.Rev.,2009,109:6765 36 Wu,X.N.;Zhao,Y.X.;Xue,W.;Wang,Z.C.;He,S.G.;Ding,X. L.Phys.Chem.Chem.Phys.,2010,12:3984
37 Espinosa-Garcia,J.;Corchado,J.C.J.Chem.Phys.,2000,112: 5731
38 Fokin,A.A.;Schreiner,P.R.Chem.Rev.,2002,102:1551
39 Dobbs,K.D.;Dixon,D.A.;Komornicki,A.J.Chem.Phys.,1993, 98:8852
40 Sander,S.P.;Friedl,R.R.;Ravishankara,A.R.;Golden,D.M.; Kolb,C.E.;Kurylo,M.J.;Huie,R.E.;Orkin,V.L.;Molina,M.J.; Moortgat,G.K.;Finlayson-Pitts,B.J.Chemical kinetics and photochemical data for use in atmospheric studies:evaluation Number 14,JPL Publication 02-25.Pasadena,CA:Jet Propulsion Laboratory,2003
