广东含笑和诗琳通含笑叶绿体基因组结构及系统发育分析
2025-01-26邓演田淑意刘世晗梁建陶家璐邓小梅




摘要:【目的】研究广东含笑(Michelia guangdongensis)和诗琳通含笑(M. sirindhorniae)的叶绿体全基因组序列,并对55种木兰科植物进行系统发育分析,为木兰科植物分类及广东含笑、诗琳通含笑遗传资源保护研究提供参考。【方法】使用高通量测序技术对广东含笑和诗琳通含笑进行叶绿体全基因组测序,获得测序结果后使用SOAPnuke 1.3.0和SPAdes 3.10.1进行组装,使用GeSeq进行基因注释, 最终获得广东含笑、诗琳通含笑叶绿体基因组序列完整的注释。注释的序列提交至GenBank,广东含笑和诗琳通含笑获得的登录号分别是MT800938、MT7876670根据注释结果,使用Chloroplot绘制叶绿体基因组结构图,用Geneious 7.1.4软件计算GC含量,使用MEGA 7.0.26检测出相对同义密码子使用频率,运用mVISTA在线软件比较叶绿体基因组序列变异,借助DnaSP 6.12.03研究基因选择压力,并基于55个木兰科叶绿体基因组中的蛋白质编码基因(protein coding gene, PCG),使用RAxML 构建了系统发育树。【结果】广东含笑、诗琳通含笑的叶绿体基因组全长分别是160 142和160 109 bp,大单拷贝区(large single-copy region, LSC)长度分别为88 170和88 137 bp,小单拷贝区(small single-copy region, SSC) 长度分别为18 822和18 810 bp,反向重复区(inverted repeat, IR)长度分别为26 575和26 581 bp。广东含笑、诗琳通含笑叶绿体基因组的总GC含量均为39.24%,成功注释基因均有112个,均包括78个蛋白质编码基因,30个tRNA基因,4个rRNA基因。【结论】对比发现广东含笑和诗琳通含笑的叶绿体基因组大小、PCT类型、结构与近源种高度相似。广东含笑、诗琳通含笑和另外53种木兰科植物种叶绿体PCG共同构建系统发育树显示,广东含笑和诗琳通含笑同在含笑属分支,首次从叶绿体基因组层面上证明诗琳通含笑属于含笑属。
关键词:广东含笑;诗琳通含笑;叶绿体基因组;濒危种;系统发育
中图分类号:S792.99;Q943""" 文献标志码:A
开放科学(资源服务)标识码(OSID):
文章编号:1000-2006(2025)01-0069-09
Analysis of chloroplast genome structures and the" phylogeny of Michelia guangdongensis and M." sirindhorniae
DENG Yanwen, TIAN Shuyi, LIU Shihan, LIANG Jian, TAO Jialu, DENG Xiaomei*
(College of Forestry and Landscape Architecture, South China Agricultural University, Guangdong Key Laboratory for Innovative Development and Utilization of Forest Plant Germplasm, Guangzhou 51064" China)
Abstract: 【Objective】This study investigated the complete chloroplast genome sequences of Michelia guangdongensis and M. sirindhorniae to explore their genomic characteristics and facilitate the comprehensive phylogenetic analysis alongside 55 other species within the family Magnoliaceae. The study aimed to provide robust genetic resource references to support the taxonomic classification of Magnoliaceae and to determine strategies for the conservation of the genetic resources of these two Michelia species, both of which hold ecological and botanical significance. 【Method】A high-throughput sequencing technology was employed to sequence the chloroplast genomes of M. guangdongensis and M. sirindhorniae. The sequencing data was processed and assembled using SOAPnuke 1.3.0 and SPAdes 3.10.1 to reconstruct the complete chloroplast genome sequences. The gene annotation was conducted using the GeSeq annotation tool, ensuring that both structural and functional gene information was comprehensively identified and recorded. This annotation process resulted in the full, detailed chloroplast genome sequences for both species. These annotated genome sequences were subsequently submitted to GenBank, with accession numbers MT800938 for M. guangdongensis and MT787667 for M. sirindhorniae. The structural analysis of the chloroplast genome was conducted using the Chloroplot visualization tool, which enabled the generation of detailed graphical representations of the genome structures. Geneious 7.1.4 was employed to calculate the GC(Guanine amp; Cytosine) content of the genomes, which serves as an important indicator of genomic stability and evolution. Relative synonymous codon usage, a metric often associated with translational efficiency and gene evolution, was analyzed using MEGA 7.0.26. mVISTA was employed for comparative genomic analysis, allowing for the exploration of sequence variations between the chloroplast genomes of these two species and other related species. Gene selection pressure, indicative of evolutionary pressures on individual genes, was analyzed using DnaSP 6.12.03. Finally, to elucidate the phylogenetic relationships within the Magnoliaceae family, a phylogenetic tree was constructed using the protein-coding sequences from the chloroplast genomes of 55 species, including M. guangdongensis and M. sirindhorniae. This analysis was performed using RAxML, which provided a maximum likelihood framework for the construction of highly accurate phylogenetic trees.【Result】The complete chloroplast genome of M. guangdongensis was found to be 160 142 base pairs (bp) in length, while that of M. sirindhorniae was slightly shorter, at 160 109 bp. Both genomes exhibited a typical quadripartite structure consisting of a large single-copy (LSC) region, a small single-copy (SSC) region, and two inverted repeat (IR) regions. Specifically, the LSC regions were 88 170" and 88 137 bp in length for M. guangdongensis and M. sirindhorniae, respectively. The SSC regions measured 18 822" and 18 810 bp, while the IR regions were nearly identical, at 26 575" and 26 581 bp. The GC content, a key indicator of genome composition and function, was found to be 39.24% for both species. Chloroplast genome annotation identified a total of 112 genes in each species. These included 78 protein-coding genes, 30 transfer RNA (tRNA) genes, and 4 ribosomal RNA (rRNA) genes. The high degree of similarity in genome size, structure, and composition between these two species underscores their close evolutionary relationship and aligns with observations made in other species within the genus Michelia. The comparative analysis using mVISTA revealed that the chloroplast genomes of M. guangdongensis and M. sirindhorniae share high levels of conservation with other members of Magnoliaceae, particularly in coding regions. However, some sequence variations were observed, predominantly in intergenic spacer regions, which can provide valuable insights into species-specific adaptations and evolutionary divergence. Selection pressure analysis using DnaSP revealed that most protein-coding genes in the chloroplast genomes were under purifying selection, consistent with the evolutionary conservation observed in chloroplast genomes across plant species. The phylogenetic tree constructed from the protein-coding genes of the chloroplast genomes provides new insights into the taxonomic positioning of M. guangdongensis and M. sirindhorniae. Both species were clustered within the same branch of the genus Michelia, confirming their close genetic relationship. Remarkably, this study marks the first instance of placing M. sirindhorniae definitively within the genus Michelia at the chloroplast genome level. This finding not only enhances our understanding of the evolutionary history of these species but also provides a more accurate framework for their taxonomic classification within Magnoliaceae. 【Conclusion】The analysis of the chloroplast genome sequences of M. guangdongensis and M. sirindhorniae significantly enhances our understanding of their genomic structure and evolutionary relationships. The observed high similarity in genome size, structure, and GC content between these two species aligns with the general characteristics of the genus Michelia. Furthermore, the phylogenetic analysis based on protein-coding genes provides compelling evidence for the placement of M. sirindhorniae within Michelia. This study enriches the genetic resource base for the family Magnoliaceae and offers valuable insights into the conservation and classification of these ecologically important species. Future studies can expand on these findings by incorporating additional molecular markers and sequencing data from nuclear genomes to further refine the taxonomic and phylogenetic frameworks for Magnoliaceae. Conservation strategies for M. guangdongensis and M. sirindhorniae should leverage the genetic insights gained from this study to ensure the sustainable preservation of their unique genetic resources in the face of habitat loss and environmental change.
Keywords:Michelia guangdongensis;Michelia sirindhorniae;chloroplast genome;endangered species;phylogeny
木兰科(Magnoliaceae)共有18属约335种植物,是现存被子植物中最原始的类群之一[1]。含笑属(Michelia)是木兰科的第2大属,在中国有41种,分布于西南部至东部[2]。诗琳通含笑(Michelia sirindhorniae)是木兰科含笑属常绿高大乔木,原产于泰国。其树干挺直,树高可达30~35 m,树形优美,枝繁叶茂,花单生,为黄色花,花香浓郁,具有较高的观赏价值。由于生境遭到破坏,诗琳通含笑群体数量日益减少,在世界自然保护联盟(IUCN)红色名录中被列为濒危(EN)物种[3]。诗琳通含笑形态同时具有木兰属与含笑属的特征,因此又名诗琳通木兰。在2005年,经华南植物园的张新华等[4]鉴定认为其应归为含笑属并为其改名。广东含笑(Michelia guangdongensis)是木兰科含笑属常绿灌木或小乔木[5],是近年来新发现的广东特有树种,其树形美观,叶片颜色独特,花美芳香,可作为优良的庭院观赏树种广泛栽植[6]。广东含笑在《中国高等植物受威胁物种名录》中被列为极危(CR)物种[7],同时也是国家二级重点保护野生植物[8]。
叶绿体(chloroplast, cp)是植物重要的细胞器,参与植物光合作用。叶绿体基因组具有较为保守的基因组序列和结构[9],可作为物种分类的依据,广泛应用于植物系统发育研究[10]。由于木兰科植物存在种间形态相似,特征重叠的情况,其分类系统存在较大的争议[11]。随着基因组测序技术的发展和研究的深入,有研究者在木兰科植物叶绿体基因组水平上开展研究,但目前鲜见关于广东含笑、诗琳通含笑叶绿体基因组的相关报道。本研究对广东含笑、诗琳通含笑的叶绿体基因组进行高通量测序,对测序结果进行组装、注释和绘图,对两物种的叶绿体基因结构特征、简单重复序列(simple sequence repeat, SSR)位点、密码子偏好性、基因组变异进行分析,并构建系统发育树,旨在为木兰科植物分类及广东含笑、诗琳通含笑遗传资源保护研究提供参考。
1 材料与方法
1.1 植物材料及其DNA提取
本研究所用广东含笑、诗琳通含笑材料采集于中国华南植物园(113°21′E,23°10′N),剪取长势较好的鲜叶运送至华南农业大学实验室,由贝纳基因公司进行测序。实验流程按照 Illumina公司提供的标准protocol执行,包括样品质量检测、文库构建、文库质量检测和文库测序等流程。具体操作如下:使用CTAB法从叶片中分离出基因组DNA[12],用机械打断的方法(超声波)将DNA片段化,随后对片段化的DNA进行片段纯化、末端修复、3′端加A等;再用琼脂糖凝胶电泳进行片段大小选择,PCR扩增形成测序文库(NEBNextUltraTMDNA Library Prep Kit for Illumina),建好的文库进行文库质检,质检合格文库用 Illumina NovaSeq进行测序。对原始测序得到的序列用SOAPnuke 1.3.0[13]软件进行过滤,过滤标准是去除 N 碱基含量超过5%的阅读子(reads)、去除低质量(质量值≤5)碱基数目达到 50%的 reads 和去除有 adapter 污染的 reads,最终得到有效数据 (clean reads)。
1.2 叶绿体全基因组的组装、注释
使用SPAdes 3.13.0(参数为-k 127)[14]进行基因组拼接,获得拼接的序列后,在NCBI (https://www.ncbi.nlm.nih.gov/) 中以溜叶含笑(M. laevifolia,NC035956)的叶绿体基因组为参考序列进行组装,使用GeSeq[15]进行基因注释,最终获得广东含笑、诗琳通含笑的叶绿体基因组序列完整的注释。注释的序列已提交至GenBank(DNA序列数据库),广东含笑登录号为MT800938,诗琳通含笑的登录号为MT787667。根据注释结果,使用Chloroplot (https://irscope.shinyapps.io/Chloroplot/)[16]绘制广东含笑、诗琳通含笑的叶绿体基因组结构图,用Geneious 7.1.4 软件(https://www.Geneious.com/)[17]计算GC含量。
1.3 基因组序列分析
使用MEGA" 7.0.26[18]基于广东含笑和诗琳通含笑的78个蛋白质编码基因(protein-coding gene, PCG)序列检测出叶绿体全基因组相对同义密码子使用度(relative synonymous codon usage, RSCU)。使用Prep-Cp (http://prep.unl.edu/)[19],进行广东含笑、诗琳通含笑叶绿体基因组序列的RNA编辑位点的预测,为提高预测的准确性,设置参数阈值(cutoff value)为0.8。使用MISA (https://webblast.ipk-gatersleben.de/misa/)在广东含笑、诗琳通含笑叶绿体基因组数据中搜索SSR位点,设置参数时,单核苷酸、二核苷酸、三核苷酸、四核苷酸、五核苷酸和六核苷酸重复,设置的最小重复数分别为8、5、4、3、3和3。
使用mVISTA在线软件(https://genome.lbl.gov/vista/mvista/instructions.shtml),以黄兰(M. champaca)叶绿体基因组(GenBank 登录号是MT800897)为参照基因组,比较5个木兰科植物种叶绿体基因组的变异,选用检测基因重排和倒位的全局比对模式(Shuffle-LAGAN)。基于5个木兰科植物种叶绿体基因组的PCG,用DnaSP 6.12.03确定非同义替换率(dN)与同义替换率(dS)的比值,以研究基因选择压力[20]。
1.4 构建系统发育树
为了研究被调查种之间的进化关系,使用RAxML[21]构建系统发育树。该系统发育树是基于55种木兰科植物叶绿体基因组中的PCG构建的,这55种木兰科植物中除广东含笑和诗琳通含笑,其余植物种的叶绿体基因组信息从GenBank上获取,Bootstrap设置为1 000。
2 结果与分析
2.1 广东含笑和诗琳通含笑的叶绿体基因组结构及特征
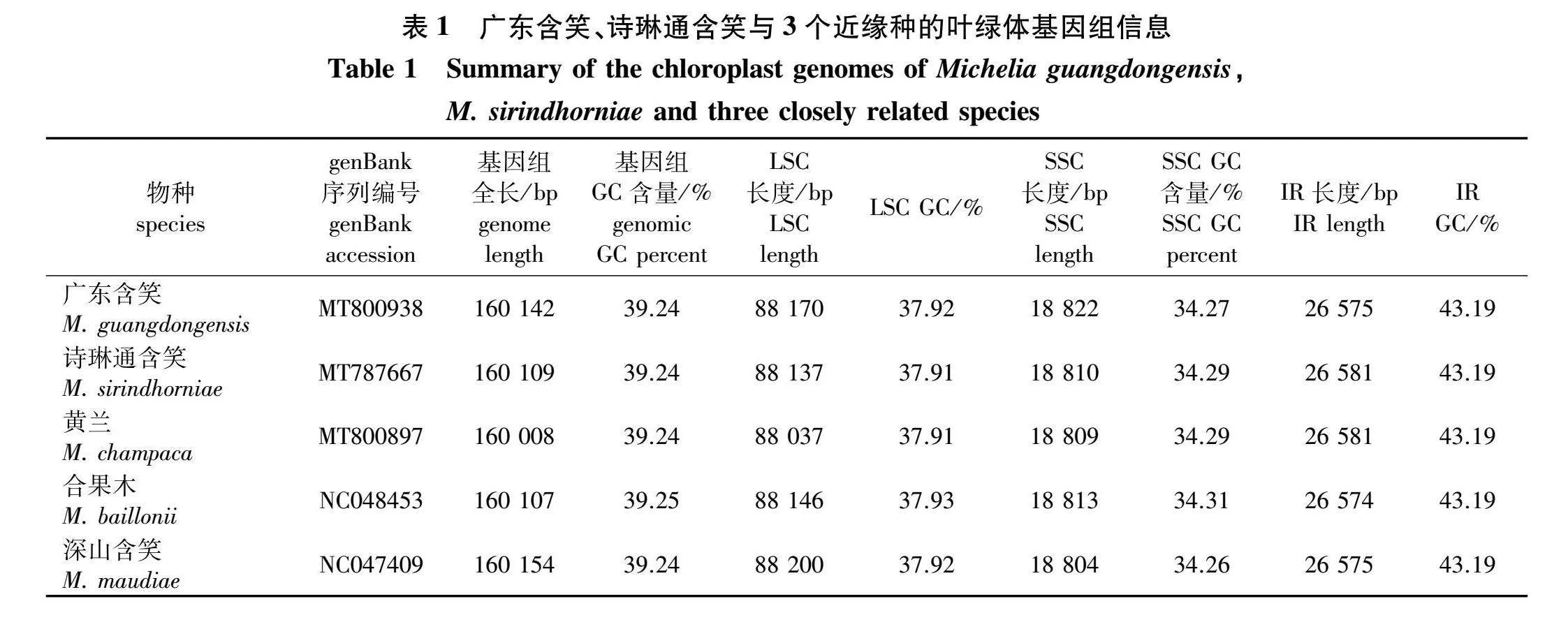
广东含笑、诗琳通含笑的叶绿体基因组均为典型的四分结构,由LSC、SSC和2个IR(IRa/IRb)组成(图1、表1)。广东含笑、诗琳通含笑的叶绿体基因组全长分别是160 142和160 109 bp,LSC长度分别为88 170和88 137 bp,SSC长度分别为18 822和18 810 bp,分割LSC、SSC的IR长度分别为26 575和26 581 bp。二者成功注释基因均有112个,均包括78个PCG,30个tRNA基因,4个rRNA基因。广东含笑、诗琳通含笑的叶绿体基因组包含的基因一致,二者的基因均被分为3大类:第I类是参与自我复制的4个核糖体RNA基因,30个转运RNA基因,12个核糖体蛋白小亚基基因,8个核糖体蛋白大亚基基因,4个RNA聚合酶基因;第Ⅱ类是参与光合作用的6个ATP合成酶基因,11个NADH脱氢酶基因,6个细胞色素b/f复合体基因,5个光系统I基因,14个光系统Ⅱ基因,1个二磷酸核酮糖羧化酶大亚基基因;第Ⅲ类是假定阅读框ycf系列基因和潜在的蛋白质编码基因(图1)。
广东含笑、诗琳通含笑叶绿体基因组的总GC含量均为39.24%,将广东含笑、诗琳通含笑的叶绿体基因组信息与黄兰、合果木(M. baillonii)、深山含笑(M. maudiae)这3种木兰科植物对比,发现这5种木兰科植物叶绿体基因组的大小、GC含量等高度相似(表1)。其中黄兰、合果木、深山含笑的叶绿体基因组从GenBank获取,登录号分别为MT800897、NC048453、NC047409。
2.2 广东含笑与诗琳通含笑的叶绿体基因组密码子使用度
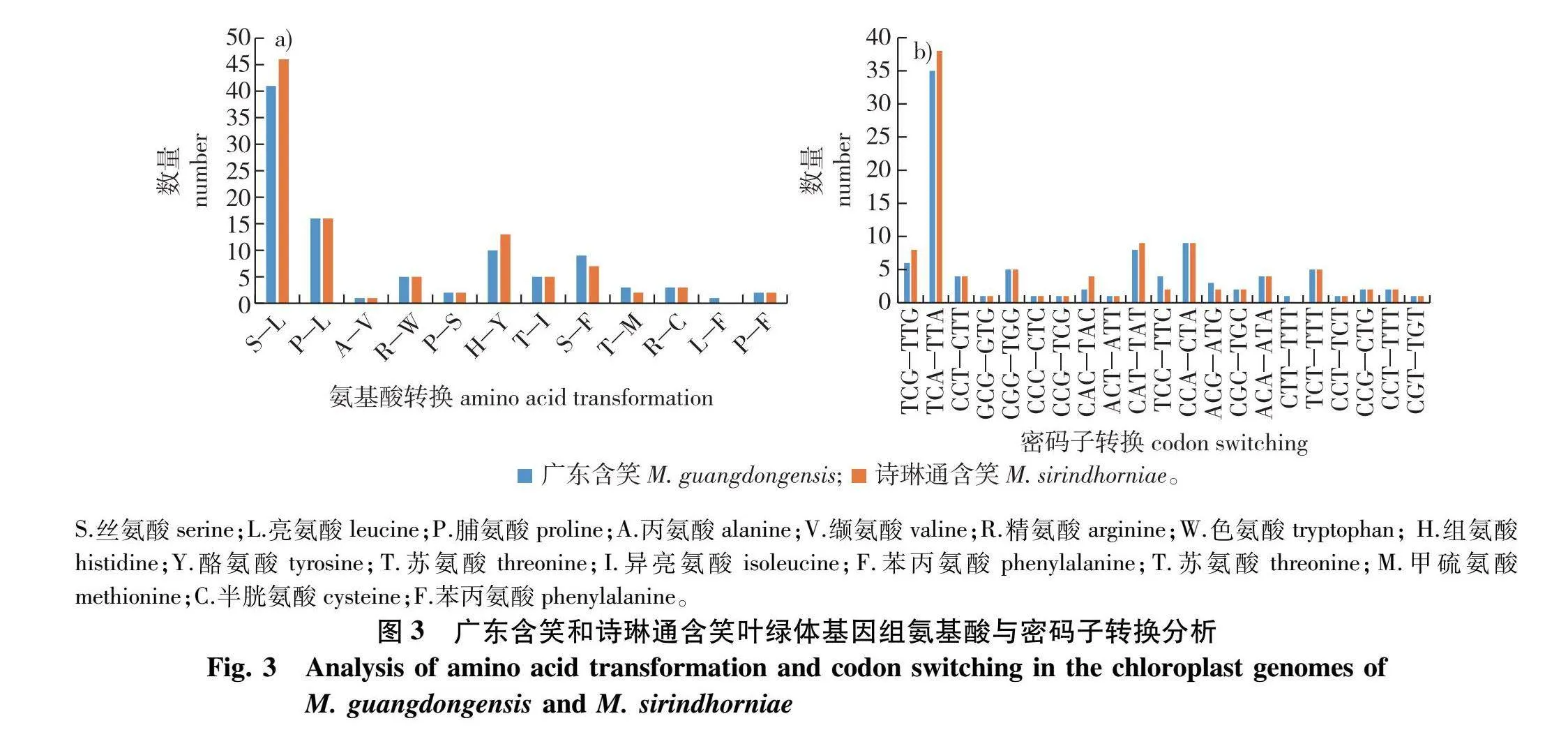
检测PCG序列,广东含笑有31个高频密码子(RSCUgt;1);诗琳通含笑有28个高频密码子(RSCUgt;1),其中密码子AGA(编码精氨酸)的RSCUgt;2。二者都有两个密码子没有密码子偏向性(RSCU=1),分别是AUG(编码蛋氨酸)和UGG (编码色氨酸) (图2)。
2.3 广东含笑与诗琳通含笑的叶绿体基因组RNA编辑
基于PCG序列分别从广东含笑和诗琳通含笑叶绿体基因组中检测到98和102个RNA编辑位点。结果表明广东含笑和诗琳通含笑可编辑的RNA大多数由丝氨酸(S)转化为亮氨酸(L)(图3a)。两个物种的密码子第3位碱基不可编辑,其中广东含笑23.5%的密码子第1位可编辑,78.6%的第2位可编辑;诗琳通含笑24.5%的密码子第1位可编辑,77.5%的第2位可编辑(图3b)。
2.4 广东含笑与诗琳通含笑的简单重复序列
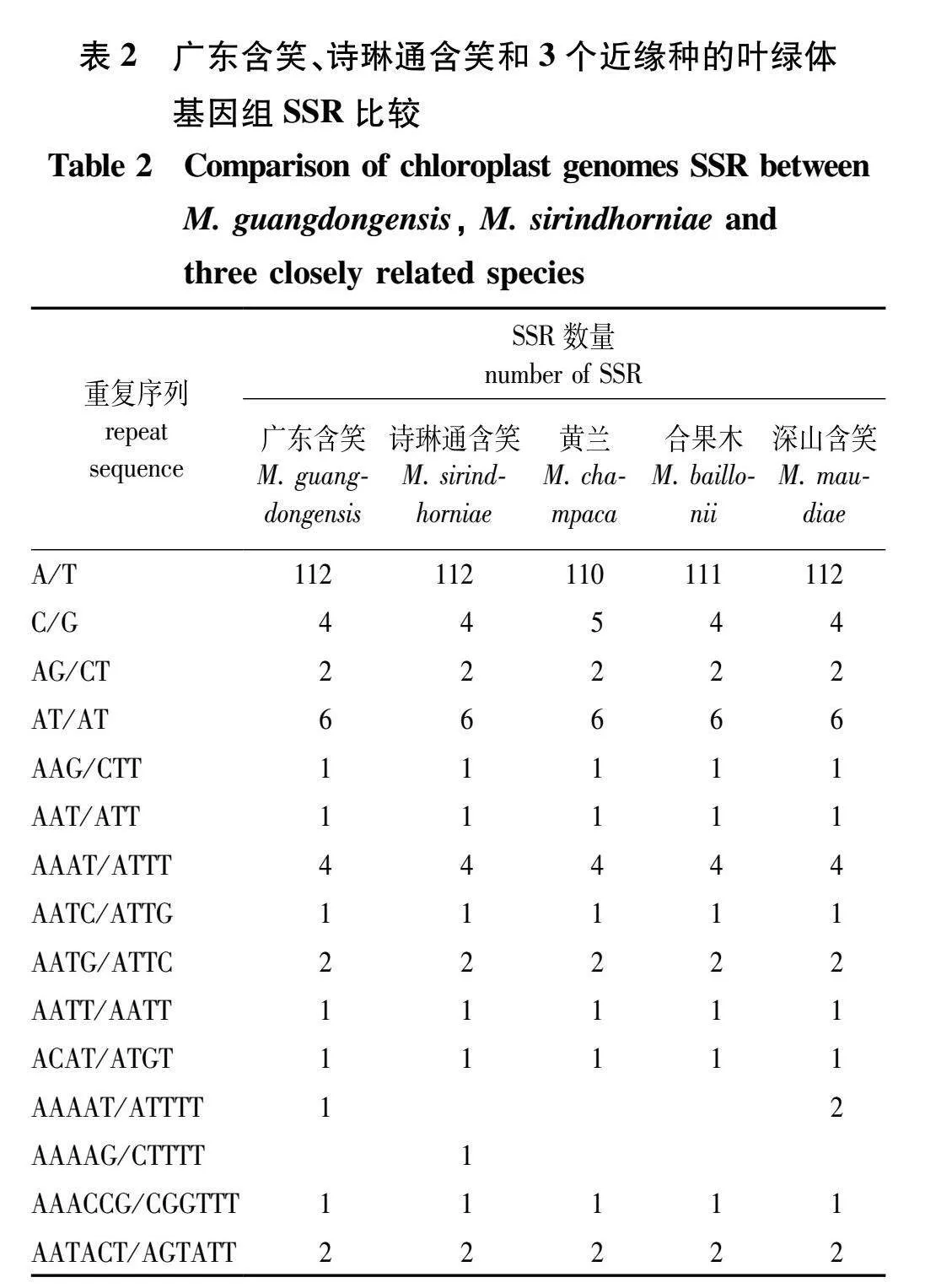
SSR广泛存在于高等植物的叶绿体基因组中,这些序列可用于分子标记,对种质资源遗传多样性分析和标记辅助选择育种具有重要的研究价值。广东含笑和诗琳通含笑叶绿体基因组均有139个SSR且类型相似。将广东含笑、诗琳通含笑的SSR和黄兰、合果木、深山含笑的进行对比,发现单核苷酸重复序列最多,数量在115~116。多种核苷酸重复序列数量一致,分别是二核苷酸重复序列(8个)、三核苷酸重复序列(2个)、四核苷酸重复序列(9个)、六核苷酸重复序列(3个)。其中黄兰和合果木没有五核苷酸重复序列(表2)。
2.5 基因序列变异分析
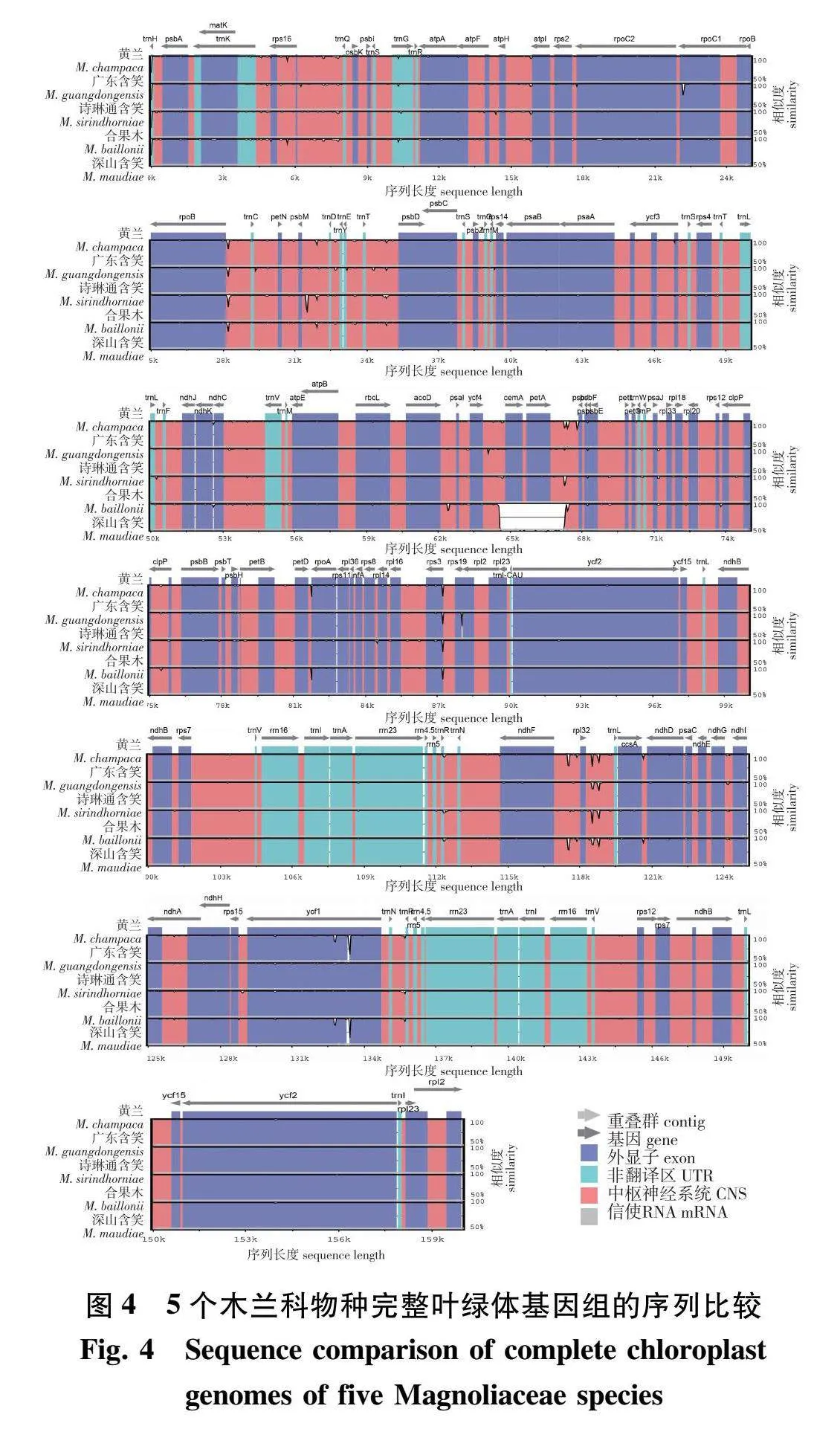
以黄兰叶绿体基因组为参照基因组,与广东含笑、诗琳通含笑、合果木和深山含笑进行基因序列变异比较,比较结果如图4所示(图中空白越大,相对应的序列差异越大)。从图4可知,这5个木兰科物种的叶绿体基因组各个部分排序顺序相对一致、高度保守。其中LSC和SSC区变异程度较高,IR区变异程度较低,非编码序列比编码序列具有更大程度的变异。在这5个物种中没有发现明显的基因插入,但深山含笑的基因cemA和petA缺失。从图4还可发现广东含笑和诗琳通含笑在matK、rpoC1、rps19、ycf1等基因有较明显空白,差异较大。
2.6 选择压力分析
检测广东含笑和诗琳通含笑叶绿体基因组中PCG的选择压力,基于广东含笑、诗琳通含笑和另外3种木兰科物种的78个PCG序列计算了非同义替换率(RdN)与同义替换率(RdS)的比值,结果得出绝大部分基因处于负选择的影响下,只有两个基因RdN/RdS的比值大于 受正选择的影响,这两个基因分别是深山含笑vs诗琳通含笑的matK、深山含笑vs黄兰的ycf1(图5)。
2.7 系统发育关系
本研究中的广东含笑、诗琳通含笑的叶绿体基因组,加上另外53种木兰科物种的叶绿体基因组共同构建系统发育树。在系统发育树中共有49个分支节点,大部分分支节点有很好的支持率(100%),其中有41个节点的支持率大于80%。根据系统发育树可知,广东含笑和诗琳通含笑同在含笑属分支,广东含笑与深山含笑关系最密切、诗琳通含笑与黄兰关系最密切(图6)。
3 讨 论
高等植物的叶绿体基因组大小多为120~160 kb,其结构与序列变异较小,都具有高度保守性[22],对研究物种的进化、种间关系以及物种鉴定具有重要意义。本研究展示了广东含笑和诗琳通含笑完整的叶绿体基因组信息,广东含笑是极危物种,诗琳通含笑是濒危物种,均具有较高的研究价值。
广东含笑的叶绿体基因组碱基数量比诗琳通含笑多33 bp,检测广东含笑、诗琳通含笑叶绿体基因组的PCG发现,广东含笑比诗琳通含笑多3个高频密码子,其中诗琳通含笑的密码子AGA使用频率最高(RSCUgt;2),广东含笑没有RSCUgt;2的密码子。相比诗琳通含笑,广东含笑叶绿体基因组RNA可编辑的密码子由CTT转换为TTT,RNA编辑后亮氨酸转化为苯丙氨酸,RNA编辑是转录后的RNA在编码区发生碱基的加入、丢失或转换等现象,广东含笑和诗琳通含笑的RNA编辑都是从胞嘧啶转换成尿嘧啶,主要发生在密码子的第2位,其次是第1位,密码子第3位没有发生RNA编辑,与大多数植物叶绿体RNA编辑位点偏好表现一致[19]。广东含笑和诗琳通含笑中发生RNA编辑后大多数转化是从极性基团变为非极性基团,由亲水性氨基酸向疏水性氨基酸转变,二者的RNA编辑都能提高蛋白质的稳定性。比较广东含笑和诗琳通含笑的叶绿体基因组SSR,广东含笑有AAAAT/ATTTT的重复序列,没有AAAAG/CTTTT的重复序列,而诗琳通含笑则相反;将广东含笑和诗琳通含笑的叶绿体基因组与另外3个木兰科植物种作比较,其SSR的区别主要集中在AAAAT/ATTTT、AAAAG/CTTTT这两个重复序列上;SSR是由1~6个核苷酸组成的短串联DNA重复序列,这些序列可用于分子标记,对种质资源遗传多样性分析和标记辅助选择育种具有重要的研究价值[23]。广东含笑、诗琳通含笑的叶绿体基因组信息,可为进一步分析2种含笑的遗传多样性和差异性提供理论基础。
通过比较5种木兰科植物的叶绿体基因组的序列,可看出这5种植物的叶绿体基因组具有较高保守性,没有检测到倒位或基因重排,非编码序列比编码序列具有更大程度的变异,编码区差异性较小,基因保守性则会较高[24]。广东含笑和诗琳通含笑在matK、rpoC1、rps19、ycf1等基因有较大差异,这些可以为开发新的DNA条形码提供参考。Maturase kinase(matK)基因位于叶绿体trnK基因中,是编码唯一一种与陆地植物叶绿体Ⅱ类内含子剪接相关的成熟酶,被认为是叶绿体基因组中进化速度最快的基因之一,可作为标记基因,在植物物种进化和鉴定等相关领域起重要作用[25]。在叶绿体基因组PCG的选择压力分析上发现,深山含笑vs诗琳通含笑的matK基因存在正向选择,由此可推测matK序列可能作为DNA 条形码区分深山含笑和诗琳通含笑。现已有研究发现桉树具有耐盐碱性,在盐胁迫环境下,随着盐碱浓度的升高,matK基因表达应激性表现出先上升后下降的趋势[26],推测诗琳通含笑在matK基因正向选择进化的影响下,可能具有一定的耐盐碱能力,可进行进一步实验验证。
木兰科植物除鹅掌楸属(Liriodendron)之外,其他属的分类一直存在较高的争议,在准确分类方面存在一些困难[27],目前对木兰科的分类研究大多是基于形态学,缺少分子层面的研究分析[11]。叶绿体基因组包含足够的信息,并且已被证明叶绿体基因组能有效地阐明植物低水平系统的发育关系[1 28-29]。对木兰科植物的叶绿体基因组及其系统发育关系进行研究,有利于木兰科植物的资源筛选、鉴定和保护。本研究选取了55种木兰科植物,是目前基于木兰科植物种叶绿体基因组研究选取数量最多的,用这55种木兰科植物的叶绿体PCG构建系统发育树,结果表明广东含笑和诗琳通含笑均是含笑属物种。明确种、属的界限,将对广东含笑、诗琳通含笑的保护提供科学依据。诗琳通含笑的分类有较多争议,此前已有研究从核基因组染色体数目上分析将其归为含笑属[8],本研究的叶绿体基因组系统发育树为诗琳通含笑归为含笑属提供进一步证明。
根据传统的形态学分类,红花木莲(Manglietia insignis)属于木莲属。然而,基于叶绿体基因组的系统发育树表明,红花木莲位于含笑属进化支中,这个结果不同于传统的形态学分类和基于核基因序列的分析结果[30],因此系统发育的结论不仅考虑完整的叶绿体基因组数据,还可能需要考虑外形特征和核基因组中的某些特征来下定论。合果木是否独立成属存在争议已久,刘玉壶[31]认为合果木应单独为1个属,Nooteboom[32]认为合果木属应并入含笑属,系统发育树显示合果木位于含笑属的分支中;经典的分类系统中,木兰属包括木兰亚属和玉兰亚属,黄丽峰[33]用RAPD和ISSR这两种分子标记技术进行研究,结果赞成木兰属分为木兰亚属和玉兰亚属,本研究系统发育树的玉兰属即为木兰属分类中的玉兰亚属。木兰属的荷花玉兰(Magnolia grandiflora)、危地马拉木兰(Magnolia guatemalensis)、西康天女花(Magnolia wilsonii)在系统发育树中位于木莲属的分支中,基于叶绿体基因组PCG建立的系统发育树结果显示,木莲属与木兰属之间存在交叉现象。木兰科植物最早起源于盖裂木属植物,经过进化分成合果木属、焕镛木属(Woonyoungia)、鹅掌楸属、含笑属、木兰属等几个属,本研究的系统发育分析是基于叶绿体基因组的PCG,其结果显示木兰属的绢毛木兰(Lirianthe albosericea)、山木兰(L. delavayi)位于盖裂木属(Talauma)的分支,这可能是这些物种在进化过程中,其叶绿体基因组进化较缓慢导致的。
随着测序成本的降低以及序列组装技术的不断发展, 将有更多木兰科植物叶绿体全基因组被测序,木兰科植物的进化关系确定将可通过参考叶绿体基因组数据的系统分析得以解决。
参考文献(reference):
[1]褚可龙,马以菘,顾宝龙,等.木兰科植物引种与筛选[J].上海交通大学学报(农业科学版),2007,25(3):307-311.CHU K L,MA Y S,GU B L,et al.Transplant and selection of Magnoliaceae plants in Shanghai[J].J Shanghai Jiao Tong Univ (Agric Sci),2007,25(3):307-311.DOI: 10.3969/j.issn.1671-9964.2007.03.024.
[2]宦智群,徐小蓉,耿兴敏,等.木兰科植物组织培养技术研究进展[J].广西植物,202 42(11):1980-1993.HUAN Z Q,XU X R,GENG X M,et al.Advances in tissue culture techniques of Magnoliaceae[J].Guihaia,202 42(11):1980-1993.DOI: 10.11931/guihaia.gxzw202101048.
[3]CUI Y Y,DENG Y W,ZHENG K Y,et al.An efficient micropropagation protocol for an endangered ornamental tree species (Magnolia sirindhorniae Noot.amp; Chalermglin) and assessment of genetic uniformity through DNA markers[J].Sci Rep,2019,9(1):9634.DOI: 10.1038/s41598-019-46050-w.
[4]张新华,夏念和.木兰科植物染色体数目报道[J].热带亚热带植物学报,2005,13(6):516-518.ZHANG X H,XIA N H.Chromosome numbers of five species and one hybrid in Magnoliaceae[J].J Trop Subtrop Bot,2005,13(6):516-518.DOI: 10.3969/j.issn.1005-3395.2005.06.011.
[5]杨科明,陈新兰.广东含笑的引种繁育与园林应用研究[J].广东园林,201 33(1):44-46.YANG K M,CHEN X L.Introduction,breeding and landscape application of Michelia guangdongensis[J].Guangdong Landsc Archit,201 33(1):44-46.DOI: 10.3969/j.issn.1671-2641.2011.01.011.
[6]徐斌,朱报著,潘文,等.广东含笑的光响应特性及其最适模型研究[J].林业科学研究,2017,30(4):604-609.XU B,ZHU B Z,PAN W,et al.Photosynthetic light response characteristics of Michelia guangdongensis and practicability of six models[J].For Res,2017,30(4):604-609.DOI: 10.13275/j.cnki.lykxyj.2017.04.010.
[7]覃海宁,杨永,董仕勇,等.中国高等植物受威胁物种名录[J].生物多样性,2017,25(7):696-744.QIN H N,YANG Y,DONG S Y,et al.Threatened species list of China’s higher plants[J].Biodivers Sci,2017,25(7):696-744.DOI: 10.17520/biods.2017144.
[8]杨蕾蕾,王文广,郎校安,等.极小种群广东含笑野外资源现状[J].生物多样性,2019,27(9):1016-1020.YANG L L,WANG W G,LANG X A,et al.Resource status of Michelia guangdongensis(Magnoliaceae),a wild plant species with extremely small populations[J].Biodivers Sci,2019,27(9):1016-1020.DOI: 10.17520/biods.2019159.
[9]ZOSCHKE R,BOCK R.Chloroplast translation:structural and functional organization,operational control,and regulation[J].Plant Cell,2018,30(4):745-770.DOI: 10.1105/tpc.18.00016.
[10]季凯凯,宋希强,陈春国,等.木兰科叶绿体基因组的密码子使用特征分析[J].中国农业科技导报,2020,22(11):52-62.JI K K,SONG X Q,CHEN C G,et al.Codon usage profiling of chloroplast genome in Magnoliaceae[J].J Agric Sci Technol,2020,22(11):52-62.DOI: 10.13304/j.nykjdb.2019.0937.
[11]朱斌,钱方,王晓双,等.基于叶绿体基因组的木兰科植物系统发育探究[J].生物学杂志,202 39(3):53-58.ZHU B,QIAN F,WANG X S,et al.The phylogeny of Magnoliaceae based on chloroplast genome[J].J Biol,202 39(3):53-58.DOI: 10.3969/j.issn.2095-1736.2022.03.053.
[12]DENG Y W,LUO Y Y,HE Y,et al.Complete chloroplast genome of Michelia shiluensis and a comparative analysis with four Magnoliaceae species[J].Forests,2020,11(3):267.DOI: 10.3390/f11030267.
[13]CHEN Y X,CHEN Y S,SHI C M,et al.SOAPnuke:a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data[J].Gigascience,2018,7(1):gix120.DOI: 10.1093/gigascience/gix120.
[14]BANKEVICH A,NURK S,ANTIPOV D,et al.SPAdes:a new genome assembly algorithm and its applications to single-cell sequencing[J].J Comput Biol,201 19(5):455-477.DOI: 10.1089/cmb.2012.0021.
[15]TILLICH M,LEHWARK P,PELLIZZER T,et al.GeSeq-versatile and accurate annotation of organelle genomes[J].Nucleic Acids Res,2017,45(1):6-11.DOI: 10.1093/nar/gkx391.
[16]ZHENG S Y,POCZAI P,HYVNEN J,et al.Chloroplot:an online program for the versatile plotting of organelle genomes[J].Front Genet,2020,11:576124.DOI: 10.3389/fgene.2020.576124.
[17]孙孟涛,张峻鑫,黄体冉,等.虎杖叶绿体基因组结构与变异分析[J].生物工程学报,202 38(5):1953-1964.SUN M T,ZHANG J X,HUANG T R,et al.Genome structure and variation of Reynoutria japonica Houtt.chloroplast genome[J].Chin J Biotechnol,202 38(5):1953-1964.DOI: 10.13345/j.cjb.210843.
[18]KUMAR S,STECHER G,TAMURA K.MEGA7:molecular evolutionary genetics analysis version 7.0 for bigger datasets[J].Mol Biol Evol,2016,33(7):1870-1874.DOI: 10.1093/molbev/msw054.
[19]李泳潭,张军,黄亚丽,等.杜梨叶绿体基因组分析[J].园艺学报,2020,47(6):1021-1032.LI Y T,ZHANG J,HUANG Y L, et al.Analysis of chloroplast genome of Pyrus betulaefolia[J].Acta Hortic Sin,2020,47(6):1021-1032.DOI: 10.16420/j.issn.0513-353x.2019-0658.
[20]ROZAS J,FERRER-MATA A,SNCHEZ-DELBARRIO J C, et al.DnaSP 6:DNA sequence polymorphism analysis of large data sets[J].Mol Biol Evol,2017,34(12):3299-3302.DOI: 10.1093/molbev/msx248.
[21]STAMATAKIS A.RAxML version 8:a tool for phylogenetic analysis and post-analysis of large phylogenies[J].Bioinformatics,2014,30(9):1312-1313.DOI: 10.1093/bioinformatics/btu033.
[22]SERRANO M,WANG B J,ARYAL B,et al.Export of salicylic acid from the chloroplast requires the multidrug and toxin extrusion-like transporter EDS5[J].Plant Physiol,2013,162(4):1815-1821.DOI: 10.1104/pp.113.218156.
[23]ZHU H Y,CHOI H K,COOK D R,et al.Bridging model and crop legumes through comparative genomics[J].Plant Physiol,2005,137(4):1189-1196.DOI: 10.1104/pp.104.058891.
[24]李述成,郭生虎,贝盏临.小檗属植物叶绿体基因组序列结构及系统发育分析[J].中草药,202 53(3):818-826.LI S C,GUO S H,BEI Z L.Chloroplast genome sequences and phylogenetic analysis of Berberis genus[J].Chin Tradit Herb Drugs,202 53(3):818-826.DOI: 10.7501/j.issn.0253-2670.2022.03.022.
[25]OZDILEK A,CENGEL B,KANDEMIR G,et al.Molecular phylogeny of relict-endemic Liquidambar orientalis Mill based on sequence diversity of the chloroplast-encoded matK gene[J].Plant Syst Evol,201 298(2):337-349.DOI: 10.1007/s00606-011-0548-6.
[26]黄永健,蔡冠龙,潘东阳,等.盐胁迫对桉树4个叶绿体基因表达的影响[J].东北林业大学学报,2019,47(10):12-15.HUANG Y J,CAI G L,PAN D Y,et al.Effects of salt stress on expression of four chloroplast genes in Eucalyptus[J].J Northeast For Univ,2019,47(10):12-15.DOI: 10.13759/j.cnki.dlxb.2019.10.003.
[27]AZUMA H,GARC A-FRANCO J G,RICO-GRAY V,et al. Molecular phylogeny of the Magnoliaceae:the biogeography of tropical and temperate disjunctions[J].Am J Bot,200 88(12):2275-2285.
[28]DONG W P,XU C,LI W Q,et al.Phylogenetic resolution in Juglans based on complete chloroplast genomes and nuclear DNA sequences[J].Front Plant Sci,2017,8:1148.DOI: 10.3389/fpls.2017.01148.
[29]ZHAO M L,SONG Y,NI J,et al.Comparative chloroplast genomics and phylogenetics of nine Lindera species (Lauraceae)[J].Sci Rep,2018,8(1):8844.DOI: 10.1038/s41598-018-27090-0.
[30]NIE Z L,WEN J,AZUMA H,et al.Phylogenetic and biogeographic complexity of Magnoliaceae in the northern Hemisphere inferred from three nuclear data sets[J].Mol Phylogenet Evol,2008,48(3):1027-1040.DOI: 10.1016/j.ympev.2008.06.004.
[31]刘玉壶. 木兰科分类系统的初步研究[J]. 中国科学院大学学报, 1984, 22(2): 89-109.LIU Y H. A preliminary study on the taxonomy of the family Magnoliaceae[J].Journal of University of Chinese Academy of Sciences, 1984, 22(2): 89-109.
[32]NOOTEBOOM H P.Notes on Magnoliaceae,with a revision of Pachylarnax and Elmerrillia and the Malesian species of Manglietia and Michelia[J].Blumea,1985,31:65-121.
[33]黄丽峰.木兰科6属20种植物的RAPD和ISSR分析[D].福州:福建师范大学,2007.HUANG L F.RAPD and ISSR analysis of 20 species in 6 genera of Magnoliaceae[D].Fuzhou:Fujian Normal University,2007.
(责任编辑 吴祝华)
