基于网络药理学探究黄芩苷抗骨肉瘤的作用机制及其与SP600125协同效应的研究
2024-12-09廖兴华庞河吴航魏波




〔摘要〕 目的 本研究通过网络药理学和体外实验探讨黄芩苷治疗骨肉瘤的潜在靶点和作用机制以及其与SP600125协同效应的研究。方法 运用PharmMapper、Swiss Target Prediction、 Genecards、OMIM和TTD数据库检索黄芩苷和骨肉瘤的潜在靶点。通过STRING平台构建蛋白质-蛋白质相互作用网络图和筛选核心靶点。使用R语言进行GO和KEGG富集分析。在GEO数据库筛选出骨肉瘤中的差异表达基因。采用分子对接方法评估其与核心靶点的结合潜力。利用CCK-8细胞增殖实验、细胞迁移实验、细胞凋亡检测、透射电子显微镜和Western blot验证其作用机制。结果 通过数据库确定46个黄芩苷治疗骨肉瘤的潜在靶点。通过蛋白质-蛋白质相互作用网络筛选出23个核心靶点。GO和KEGG富集分析表明,磷脂酰肌醇三激酶(phosphatidylinositol 3-kinase, PI3K)/蛋白激酶B(protein kinase B, PKB/AKT)通路和丝裂原活化蛋白激酶(mitogen-activated protein kinase, MAPK)通路和凋亡通路在黄芩苷治疗骨肉瘤中起着关键作用。核心靶点与骨肉瘤差异性表达基因比对的结果发现,AKT1、热休克蛋白90α (heat shock protein 90 alpha, HSP90AA1)、膜联蛋白A5(annexin A5, ANXA5)、细胞周期检验点激酶1(checkpoint kinase 1, CHEK1)和尿激酶型纤溶酶原激活剂(urokinase-type plasminogen activator, PLAU)在骨肉瘤组织中的表达高于正常骨组织。分子对接揭示黄芩苷与AKT1、HSP90AA1、ANXA5、CHEK1和PLAU靶点有较好的结合活性。体外细胞实验表明,黄芩苷抑制HOS和143B骨肉瘤细胞的增殖和迁移,并促进细胞凋亡。此外,联合使用c-Jun氨基末端激酶(c-Jun N-terminal kinase, JNK)抑制剂SP600125进一步增强了黄芩苷的抗骨肉瘤作用。透射电子显微镜显示,黄芩苷增加骨肉瘤细胞内自噬小体的数量,但与SP600125联合时,黄芩苷诱导的自噬小体的增多会受到抑制。Western blot分析结果表明,黄芩苷抑制了AKT1蛋白的磷酸化和p-AKT/AKT表达水平,并与SP600125共处理可增强该抑制作用。另外,黄芩苷可诱导骨肉细胞内LC3-II和p62以及p-JNK/JNK的表达,但与SP600125联用会显著抑制此效应。结论 黄芩苷通过多靶点和多通路的相互作用发挥抗骨肉瘤效应。此外,SP600125协同增强了黄芩苷治疗骨肉瘤的效应,为骨肉瘤的治疗提供了研究依据和理论支持。
〔关键词〕 骨肉瘤;黄芩苷;网络药理学; 蛋白激酶B1;自噬;SP600125
〔中图分类号〕R285.5 〔文献标志码〕A 〔文章编号〕doi:10.3969/j.issn.1674-070X.2024.10.018
Mechanism of action of baicalin against osteosarcoma and its synergistic effects with SP600125 based on network pharmacology
LIAO Xinghua, PANG He, WU Hang, WEI Bo*
Orthopedic Center, Hospital of Guangdong Medical University, Zhanjiang, Guangdong 524001, China
〔Abstract〕 Objective To investigate the potential targets and mechanism of action of baicalin in treating osteosarcoma and its synergistic effects with SP600125 by network pharmacology and in vitro experimentation. Methods Using PharmMapper, Swiss Target Prediction, Genecards, OMIM, and TTD databases, potential targets for baicalin and osteosarcoma were retrieved. A protein-protein interaction network was constructed and core targets were screened through the STRING platform. GO and KEGG enrichment analyses were performed using R language. Differentially expressed genes in osteosarcoma were identified from the GEO database. Molecular docking was employed to assess the binding potential with the core targets. The mechanism of action was validated through CCK8 cell proliferation assays, cell migration assays, apoptosis detection, transmission electron microscopy, and Western blot. Results Through database analysis, 46 potential targets for baicalin in the treatment of osteosarcoma were identified. Among these, 23 core targets were screened via the protein-protein interaction network. GO and KEGG enrichment analyses indicated that the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB/AKT) pathway, the mitogen-activated protein kinase (MAPK) pathway, and the apoptosis pathway play pivotal roles in the treatment of osteosarcoma with baicalin. By comparing the core targets with differentially expressed genes in osteosarcoma, it was found that AKT1, heat shock protein 90 alpha (HSP90AA1), annexin A5 (ANXA5), checkpoint kinase 1 (CHK1), and urokinase-type plasminogen activator (PLAU) were expressed at higher levels in osteosarcoma tissues than in normal bone tissues. Molecular docking revealed that baicalin exhibited good binding activity with AKT1, HSP90AA1, ANXA5, CHK1, and PLAU targets. In vitro cell experiments demonstrated that baicalin inhibited the proliferation and migration of HOS and 143B osteosarcoma cells while promoting cell apoptosis. Furthermore, the combination with the c-Jun N-terminal kinase (JNK) inhibitor SP600125 further enhanced the anti-osteosarcoma effects of baicalin. Transmission electron microscopy showed that baicalin increased the number of autophagosomes within osteosarcoma cells, but this increase was inhibited when combined with SP600125. Western blot analysis indicated that baicalin inhibited the phosphorylation of AKT1 protein and reduced the p-AKT/AKT expression level, and cotreatment with SP600125 enhanced this inhibitory effect. Additionally, baicalin induced the expression of LC3-II, p62, and p-JNK/JNK in osteosarcoma cells, but this induction was significantly suppressed when combined with SP600125. Conclusion Baicalin exerts anti-osteosarcoma effects through interaction with multiple targets and pathways. Furthermore, SP600125 synergistically enhances the therapeutic efficacy of baicalin in treating osteosarcoma, providing research evidence and theoretical support for the treatment of this disease.
〔Keywords〕 osteosarcoma; baicalin; network pharmacology; protein kinase B1; autophagy; SP600125
骨肉瘤是一种常见的原发性恶性骨肿瘤,好发于青少年,主要发生四肢长骨干骺端,如股骨远端和胫骨近端[1]。骨肉瘤以其高度的恶性特征而著称,具有快速的生长速度和转移能力,因此治疗起来相当困难,导致患者的致残率和死亡率较高[2]。据统计,骨肉瘤患者的综合5年生存率仅为60%~70%,而一旦出现远处转移或复发的情况,这一比例会骤降至20%,其中转移性病灶的存在是预后不良的主要因素[3]。在初次诊断时,已有80%的骨肉瘤患者体内存在可测量的转移性或微转移性病灶。因此,几乎所有患者在接受手术切除治疗后,还需要进行新辅助化学治疗[4-5]。目前,大剂量甲氨蝶呤联合阿霉素和顺铂的化疗方案已成为治疗骨肉瘤标准化学治疗方案。然而,由于其毒副作用,在临床应用受到一定的限制[6]。此外,对于已发生转移、复发或不可切除的肿瘤患者通常会对当前化学治疗方案产生耐药[7]。因此,寻找有效的新型抗骨肉瘤药物具有重要意义。
植物天然有效成分,因其多靶点和多途径的协同作用,在肿瘤发生、发展、转移以及免疫调节等各个阶段都展现出治疗潜力,成为抗肿瘤先导化合物的重要来源[8]。黄芩苷(Baicalin,C21H18O11)是一种从黄芩植物中提取的黄酮类化合物,具备多种药理活性,包括抗氧化、抗肿瘤、抗菌和保护心脑血管功能等[9]。在抗肿瘤方面,黄芩苷表现出显著效果,并且对健康组织无显示毒性[9-10]。SP600125(C14H8N2O)是一种具有强大渗透能力的小分子化合物,它通过ATP竞争机制与JNK结合,有效地抑制了JNK1、JNK2和JNK3的活性,从而干扰了细胞的生物过程[11]。体内外的研究揭示,该化合物对多种肿瘤表现显著的治疗潜力,特别是对膀胱癌、骨肉瘤和肺癌等,其能够通过影响肿瘤细胞周期阻滞、抑制转移行为、促进凋亡和抑制自噬,以及增强肿瘤细胞对药物的敏感性来发挥作用[12]。
1 网络药理学方法
1.1 网络药理学数据库及软件
PubChem数据库(https://pubchem.ncbi.nlm.nih.gov/),PharmMapper数据库(http://www.lilab-ecust.cn/pharmmapper/),Swiss Target Prediction数据库(http://www.swisstargetprediction.ch/),GeneCards数据库(https://www.genecards.org/),OMIM数据库(https://omim.org/),TTD数据库(http://db.idrblab.net/ttd/),DAVID数据库(https://david.ncifcrf.gov/),STRING数据库(https://string-db.org/),Venny 2.1在线软件作图工具平台(https://bioinfogp.cnb.csic.es/tools/venny/),UniProt数据库(https://www.uniprot.org/),GEO数据库(https://www.ncbi.nlm.nih.gov/geo/),Cytoscape 3.8.2软件,R4.0.5软件等。
1.2 黄芩苷-骨肉瘤共同靶点的筛选
通过PubChem数据库获取其化学结构信息。随后,将此结构数据输入到PharmMapper数据库中,筛选出Norm Fit值大于0.6的潜在靶点。同时,利用Swiss Target Prediction数据库进一步预测黄芩苷可能作用的靶点。通过UniProt数据库校正所获得靶点的名称。以“Osteosarcoma”作为关键词在Gene?鄄Cards、OMIM和TTD数据库中搜索与骨肉瘤相关的靶点。收集这些疾病相关靶点的信息后,使用Venny 2.1在线工具绘制韦恩图,找出药物靶点与疾病靶点之间的交集。最终,采用Cytoscape软件构建一个“疾病-靶点-成分”网络图。
1.3 靶点相互作用网络构建及核心靶点分析
将筛选出的共同靶点输入STRING数据库中检索共同靶点之间的蛋白质-蛋白质相互作用(protein-protein interaction,PPI)网络关系。设置筛选条件为“Homo sapiens”以确保蛋白种类Human,并将相互作用阈值设定为0.4,以检索置信度较高的PPI网络数据。随后,将获得的PPI数据导入Cytoscape软件,绘制出详细的PPI网络图。接着,利用Cytoscape中的NetworkAnalyzer工具对PPI网络数据进行拓扑分析。基于Degree值参数进行排序,选取Degree值高于平均值的基因作为核心靶点。最后,使用R软件对选定的核心靶点进行数据可视化。
1.4 GO和KEGG富集分析
利用R软件结合Bioconductor生物信息学软件包,对筛选出的潜在靶点进行了基因本体论(gene ontology, GO)和京都基因与基因组数据库(Kyoto encyclopedia of genes and genomes, KEGG)的富集分析,其中设定P<0.05和Q<0.05作为筛选条件。挑选各部分GO富集分析P值排名前10的条目,通过柱状图形式呈现。富集分析基于P值排序,选出KEG富集分析的前30个条目,并采用气泡图的方式展现了涉及的信号传导通路。
1.5 核心靶点的差异表达基因分析
从GEO数据库下载了GSE99671数据集,该数据集包括18例骨肉瘤组织和18例正常骨组织的序列文件。接着,提取了骨肉瘤患者的基因表达矩阵,并对数据进行了归一化处理,将原始的Counts数据转换成TPM(transcripts per million),以实现表达量的标准化。然后利用平台注释文件对基因名进行注释。结合PPI分析确定的核心靶点与归一化后的基因表达数据,筛选出在正常样本和骨肉瘤样本之间表现出显著差异的差异表达基因。
1.6 分子对接
黄芩苷的结构来自PubChem数据库,然后导入Schr dinger软件经过加氢、结构优化和能量最小化保存后作为分子对接的配体。蛋白结构AKT1(PDB
ID:7NH5)、HSP90AA1(PDB ID:3O0I)、ANXA5(PDB ID:2XO3)、CHEK1(PDB ID:2HXL)和PLAU(PDB ID:1C5Y)来自RCSB数据库(https://www.rcsb.org/)。蛋白结构在Schr?觟dinger Maestro软件进行处理,使用Protein Preparation Wizard模块去除蛋白结晶水,补充缺失的氢原子,修复缺失的肽键和肽段信息,进行能量最小化和几何结构的优化。在Glide模块中进行筛选时,根据蛋白结构的小分子配体确定对接位点,位点盒子大小设为15 ?魡×15 ?魡×15 ?魡。最后通过Standard Precision Glide Docking方法进行分子对接和筛选。对接后的复合物使用Pymol2.1软件进行可视化,分析黄芩苷和靶点蛋白的作用模式。
2 材料
2.1 主要药品和试剂
黄芩苷(HPLC≥98%;上海源叶生物科技有限公司,批号:B20570);SP600125(HPLC≥99.7%; Medchem Express公司,批号:HY-12041);抗体p-AKT1(批号:9018)、AKT1(批号:2938)、JNK(批号:9252)、p-JNK(批号:4668)、Bax(批号:2772)、Bcl-2(批号:4223)、辣根过氧化物酶标记的山羊抗兔IgG(批号:7074)和抗小鼠IgG(批号:7076)均购买于Cell Signaling Technology公司;抗体p-AKT(批号: ab192623)、AKT(批号:ab179463)、LC3(批号:ab48394)和p62(批号:ab56416)抗体购买于Abcam公司;抗体GAPDH(批号:21612)购买于Signalway Antibody公司;1%青霉素-链霉素混合液(北京索莱宝科技有限公司,批号:P1400);10%胎牛血清FBS(批号:A5669401)、MEM基础培养基(批号:11095080)均购买于Thermo Fisher Scientific公司;ECL显色剂(Zeta Life公司,批号:310212);0.1%结晶紫(批号:C0121)、Annexin V-FITC细胞凋亡检测试剂盒(批号:C1062M)、BCA蛋白浓度测定试剂盒(批号P0010S)、5X SDS-PAGE蛋白上样缓冲液(批号:P0015)均购自碧云天生物技术研究所。
2.2 主要仪器
微孔板读取(型号:MK3,Thermo Fisher Scientific公司);流式细胞仪(型号:FACSCelesta,BD Biosciences公司);荧光成像系统(型号:Tanon-5200Multi,Tanon公司);透射电子显微镜(型号:JEM-1400,JEOL公司);超薄切片机(型号:Leica EM UC7,Leica Microsystems公司)。
3 体外细胞实验
3.1 细胞培养
将143B和HOS骨肉瘤细胞以10×105的密度接种在10 cm×10 cm的细胞培养瓶中,使用添加了1%青霉素-链霉素混合液和10%胎牛血清FBS的MEM基础培养基进行培养。细胞被放置在恒温37 ℃和5% CO2的细胞培养箱中。定期检查细胞的生长状态,并适时更换培养基。当细胞生长接近密集单层,即达到80%~90%的融合度时,便进行传代操作。
3.2 CCK-8检测细胞增殖实验
将骨肉瘤细胞悬液以每孔3.0×103个细胞的密度接种于96孔板中,并在37 ℃、5% CO2的细胞培养箱孵育24 h。之后,细胞被不同浓度(0、20、40、80 μmol·L-1)的黄芩苷或不同浓度(0、10、20、40 μmol·L-1)SP600125处理48 h。若需联合使用20 μmol·L-1浓度的SP600125,则在黄芩苷干预前对细胞进行4 h的预处理。为了评估细胞活力,向每个孔中加入10 μL
CCK-8检测溶液,在37 ℃、5% CO2的条件下继续孵育2~4 h,测定450 nm的波长的吸光度值。
3.3 细胞迁移实验
为了评估黄芩苷或SP600125对骨肉瘤细胞迁移能力的影响,使用Transwell小室进行实验。首先,将骨肉瘤细胞用MEM培养基重新悬浮,并取100 μL的细胞悬液(含2.0×104个细胞)加入Transwell小室的上室中。在下室中加入500 μL含有10% FBS的MEM培养基,然后放入恒温37 ℃、5% CO2细胞培养箱中静置培养24 h。培养结束后,轻轻用棉签擦去上室内未发生迁移的细胞。对于迁移至膜内侧的细胞,使用甲醇固定15 min,随后以0.1%结晶紫染色10 min。PBS彻底清洗掉多余的染料。最后,在放大倍数400×显微镜视野下观察染色后的细胞,并在随机选取的5个视野拍照。所得图片可通过Image J软件进行定量分析
3.4 流式细胞凋亡检测
为准确评估骨肉瘤细胞的凋亡比例,采用Annexin V-FITC细胞凋亡检测试剂检测。骨肉瘤细胞先经80 μmol·L-1黄芩苷单独处理或与20 μmol·L-1 SP600125联合处理48 h。处理后,用5 μL Annexin
V-FITC和10 μL碘化丙啶染色液对细胞进行染色,再加入195 μL Annexin V-FITC结合缓冲液重悬细胞。随后,在室温下将细胞避光孵育30 min以促进染料结合。孵育结束后,采用流式细胞仪对细胞样本进行检测,并通过BD FACSDiva软件来定量染色后的凋亡细胞比例。
3.5 透射电子显微镜观察及样品制备
前固定:骨肉瘤细胞在4 ℃下用3%戊二醛固定4 h,随后用0.1 mol·L-1二甲砷酸钠缓冲液换洗3次,每2 h一次。后固定:样本在4 ℃下用1%锇酸浸泡2 h,然后用0.1 mol/L二甲砷酸钠缓冲液冲洗2次,15 min/次。染色:室温下用饱和醋酸铀染料染色2 h。脱水与渗透:样本依次在4 ℃的50%、70%乙醇和室温的80%、90%乙醇中脱水,然后在室温下用100%乙醇处理2次,接着用丙酮渗透2次,15 min/次。最后,样本在室温下用完全包埋液(Eponate12环氧树脂)与丙酮的混合液(比例1∶1)渗透3 h,然后比例1∶2渗透3 h,最后用完全包埋液渗透过夜。包埋:样本在40 ℃下用完全包埋液浸泡12 h,然后在60 ℃条件移至包埋板并温育48 h。超薄切片:使用超薄切片机制作90 nm厚的切片。电镜观察:使用透射电子显微镜在80 kV的工作电压下观察样本,并用DigitalMicrograph软件进行图像采集。
3.6 Western bolt检测
收集经黄芩苷或与SP600125联合干预后的骨肉瘤细胞沉淀,在4 ℃条件下加入适量预混有PMSF(PMSF∶RIPA=1∶100)的RIPA裂解液。使用细胞刮刀彻底将细胞碎片收集到EP管中,并在冰上持续裂解30 min。随后进行30 s的超声处理,以促进细胞充分裂解。完成裂解后,样本在4 ℃、12 000 r/min的条件下离心(离心半径9 cm)15 min,然后将上层清澈的蛋白溶液转移到新的EP管中。根据BCA蛋白浓度测定试剂盒的说明书配制BCA定量标准曲线,并测定及调整各组蛋白样品浓度一致。向每组蛋白样品加入适量的5X SDS-PAGE蛋白上样缓冲液,混合均匀后在99 ℃下煮沸10 min使蛋白变性。使用配制好的10%或12% SDS-PAGE凝胶,每个样本孔道加载20 μg蛋白质,在100 V电压下电泳60 min进行分离。之后,在250 mA恒流条件下,将凝胶上的蛋白转印2.5 h至0.2 μm孔径的PVDF膜。PVDF膜用5%脱脂牛奶封闭60 min。根据Marker位置和目标蛋白,裁剪相应的膜片,并加入一抗p-AKT1、AKT1、JNK、p-JNK、p-AKT、AKT、Bax、Bcl-2、LC3、p62和GAPDH(均按1∶1 000稀释)在4 ℃冰箱中孵育12 h。然后与相应的二抗(均按1∶5 000稀释)在室温避光孵育1 h。最后,使用ECL显色剂结合荧光成像系统曝光膜上的蛋白条带。利用Image J软件对蛋白条带进行灰度值分析,并以GAPDH为内参计算目标蛋白的表达水平。
3.7 统计学分析
所有数据均表示为3次独立实验的“x±s”,并使用SPSS 25.0和GraphPad Prism 8.0软件进行统计分析和绘图。对于两组间的比较,采用独立样本t检验。当涉及3组及以上之间的统计分析时,则采用单因素方差分析(ANOVA)配合Tukey事后多重比较检验。P<0.05为差异具有统计学意义。
4 结果
4.1 黄芩苷治疗骨肉瘤的潜在靶点
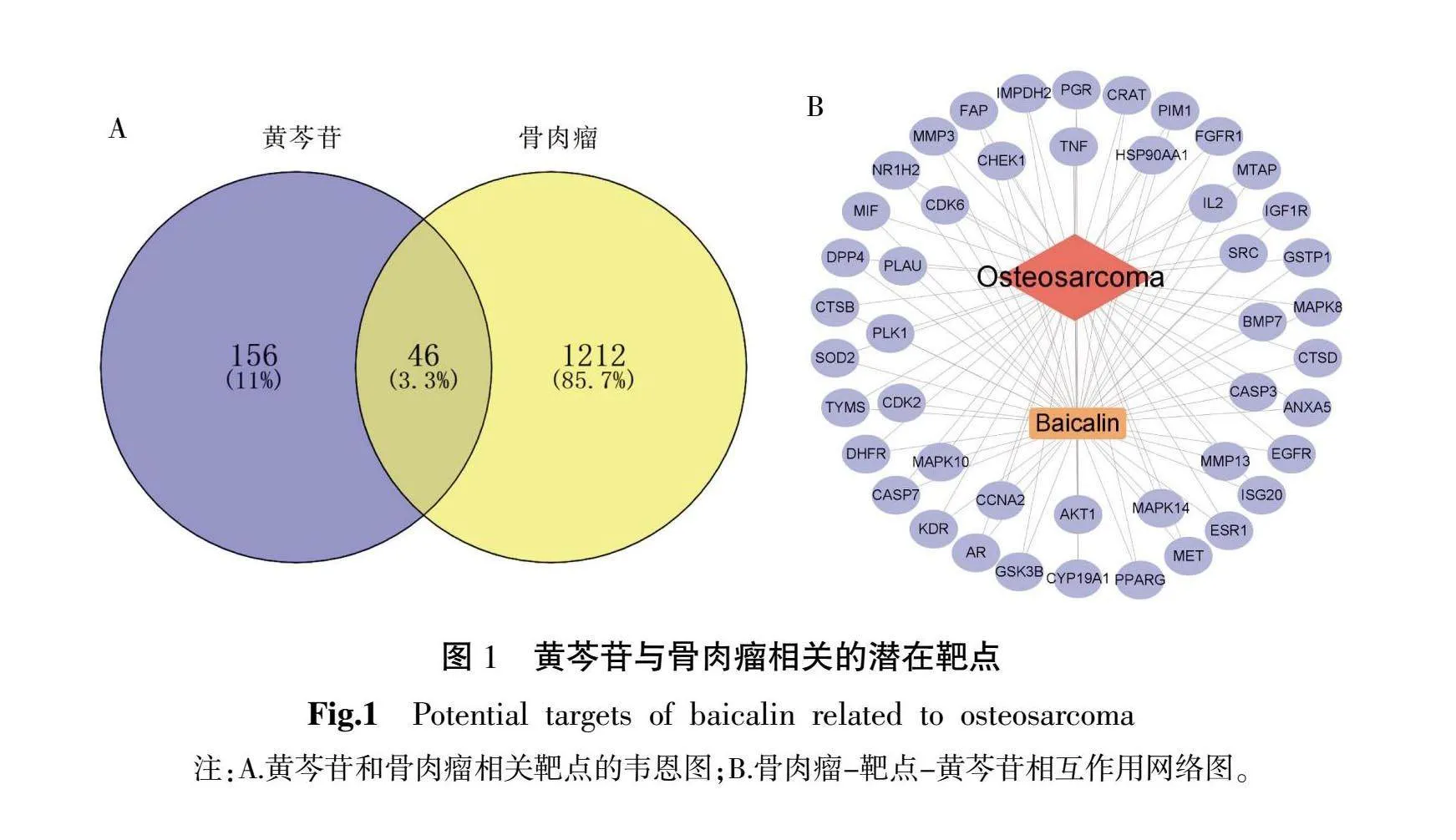
在PharmMapper和Swiss Target Prediction数据库中检索后,确定了202个黄芩苷潜在作用靶点。同时,通过Genecards、OMIM和TTD数据库的筛选,发现了1 258个与骨肉瘤疾病相关的潜在靶点。对两组数据进行交集分析,揭示了46个共同靶点(图1A)。进一步整合这些共同靶点信息,绘制了“疾病-靶点-成分”交互网络图(图1B)。
4.2 黄芩苷治疗骨肉核心靶点的筛选
通过STRING数据库检索黄芩苷治疗骨肉瘤的潜在作用数据,将数据整合并构建得到46个节点、317个边缘和3个同心圆的PPI网络图(图2A)。其中,节点的大小、颜色深浅变化根据Degree 值大小变化。PPI数据进行拓扑分析,筛取Degree值大于平均分(23个)的基因作为核心靶点(图2B)。在核心靶基因中,AKT1(36边)、CASP3(33边)、EGFR(30边)、HSP90AA1(30边)、ESR1(29边)和TNF(29边)具有较高的连接节点度。
4.3 GO和KEGG富集分析
对46个共同靶点进行GO富集分析,得到涉及生物学过程(biological process,BP)、细胞组分(cellular component,CC)和分子功能(molecular function,MF)的三大类别信息。GO分析的结果共富集到1 569条与BP相关的条目、29条与CC相关的条目以及109条与MF相关的过程。结果根据P值选取每部分排前10位的条目绘制柱状图(图3A)。KEGG富集分析筛选出111条黄芩苷治疗骨肉瘤相关的潜在通路,选取P值前30的通路绘制KEGG富集的气泡图,其中富含与癌症相关信号通路,如PI3K/AKT信号通路、MAPK信号通路以及细胞凋亡等(图3B)。
4.4 核心靶点与骨肉瘤差异性表达基因的比对
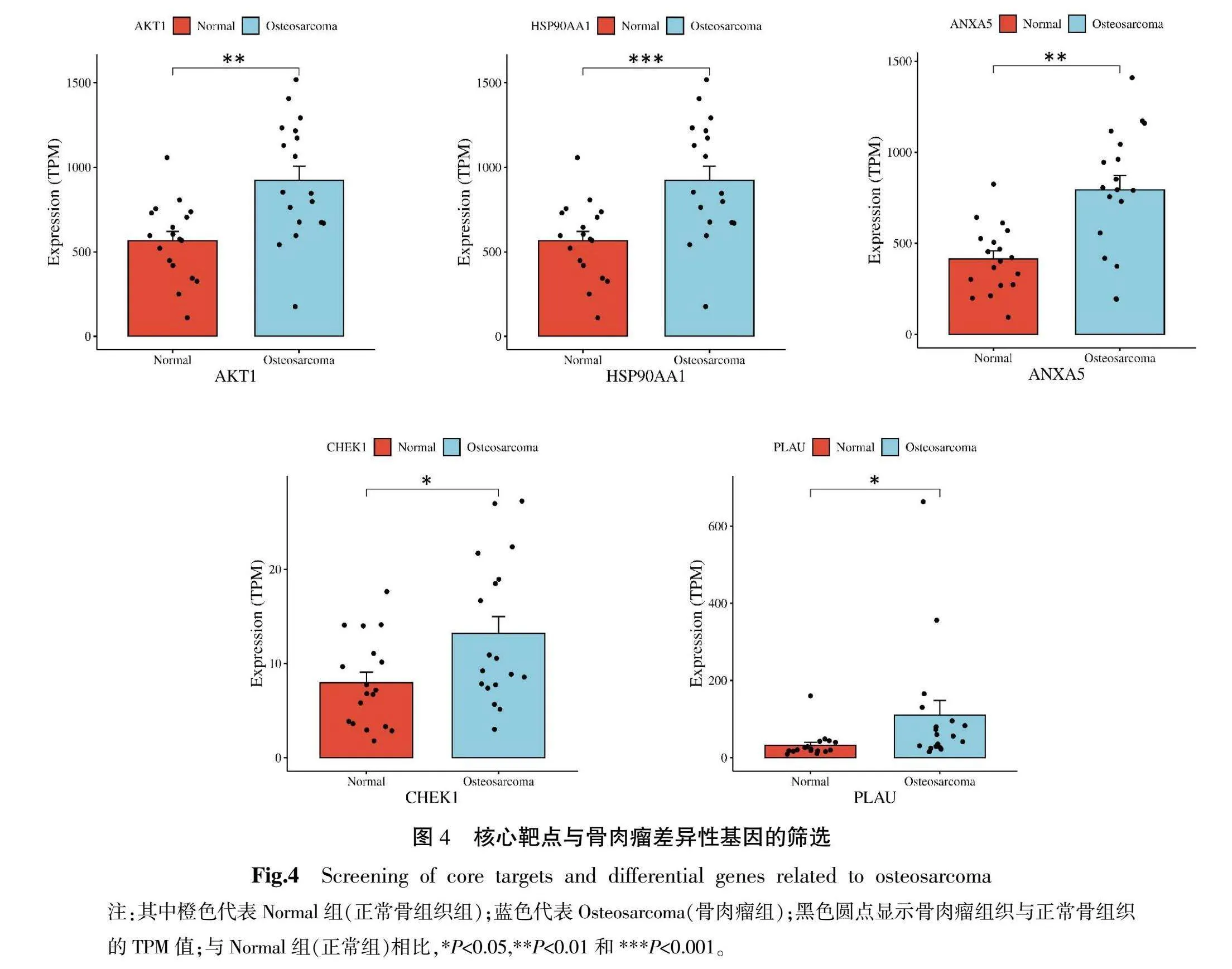
PPI的核心靶点(23个)与CEO数据库的骨肉瘤基因进行差异性表达分析。结果显示AKT1(P<0.01)、HSP90AA1(P<0.001)、ANXA5(P<0.01)、CHEK1(P<0.05)和PLAU(P<0.05)在骨肉瘤组织中的表达高于正常骨组织(图4)。然而,其他核心靶点基因,包括EGFR、CASP3、ESR1、TNF、SRC、PGR、PPARG、AR、IG?鄄F1R、MAPK14、MAPK8、CCNA2、CDK2、KDR、CDK6、IL2、MET和MMP3,在骨肉瘤和正常骨组织之间的表达差异无统计学意义(P>0.05)。
4.5 核心靶点的分子对接分析
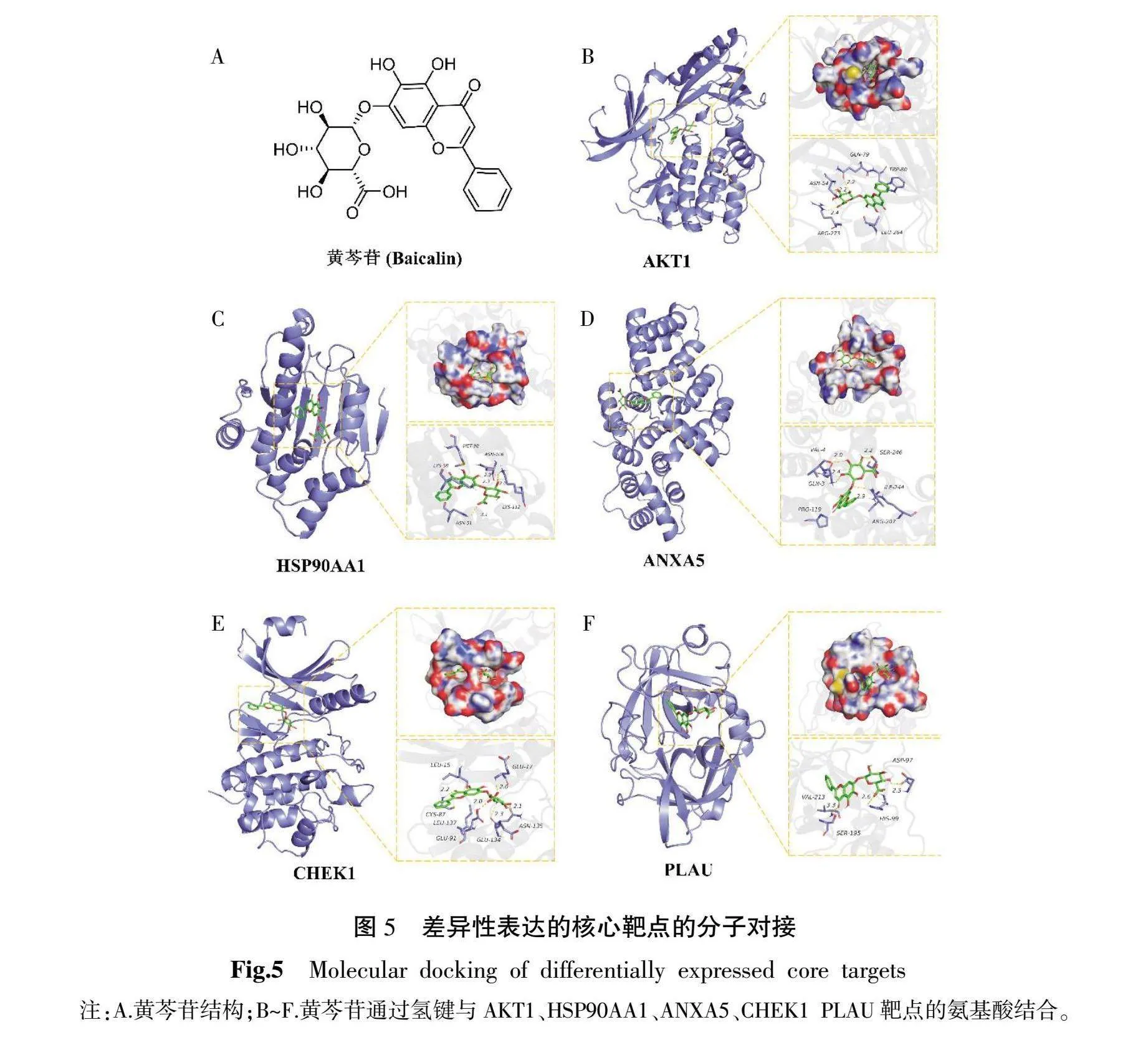
骨肉瘤差异表达的核心靶点AKT1、HSP90AA1、ANXA5、CHEK1和PLAU与黄芩苷进行分子对接,通过结合能和氢键相互作用来评价结合强度。根据结合模式清晰地显示出黄芩苷结构(图5A)与靶蛋白活性位点相互作用的氨基酸残基。结合情况如下:AKT1(结合能:-10.94 kcal/mol)活性位点的GLN-79,ASN-54和ARG-273氨基酸与黄芩苷形成氢键相互作用(图5B);HSP90AA1(结合能:-9.36 kcal/mol)活性位点的ASN-106、LYS-112、ASN-52和LYS-58氨基酸与黄芩苷形成氢键相互作用(图5C);ANXA5 (结合能:-8.62 kcal/mol)活性位点的SER-246、ILE-244、GLN-3和VAL-4氨基酸与黄芩苷形成氢键相互作用(图5D);CHEK1(结合能:-9.11 kcal/mol)活性位点的GLU-17、GLU-134、ASN-135、CYS-87和GLU-91氨基酸与黄芩苷形成氢键相互作用(图5E);PLAU(结合能:-8.64 kcal/mol)活性位点的ASP-97、HIS-99和SER-195氨基酸与黄芩苷形成氢键相互作用(图5F)。
4.6 黄芩苷对骨肉瘤细胞增殖及核心靶点AKT1的影响
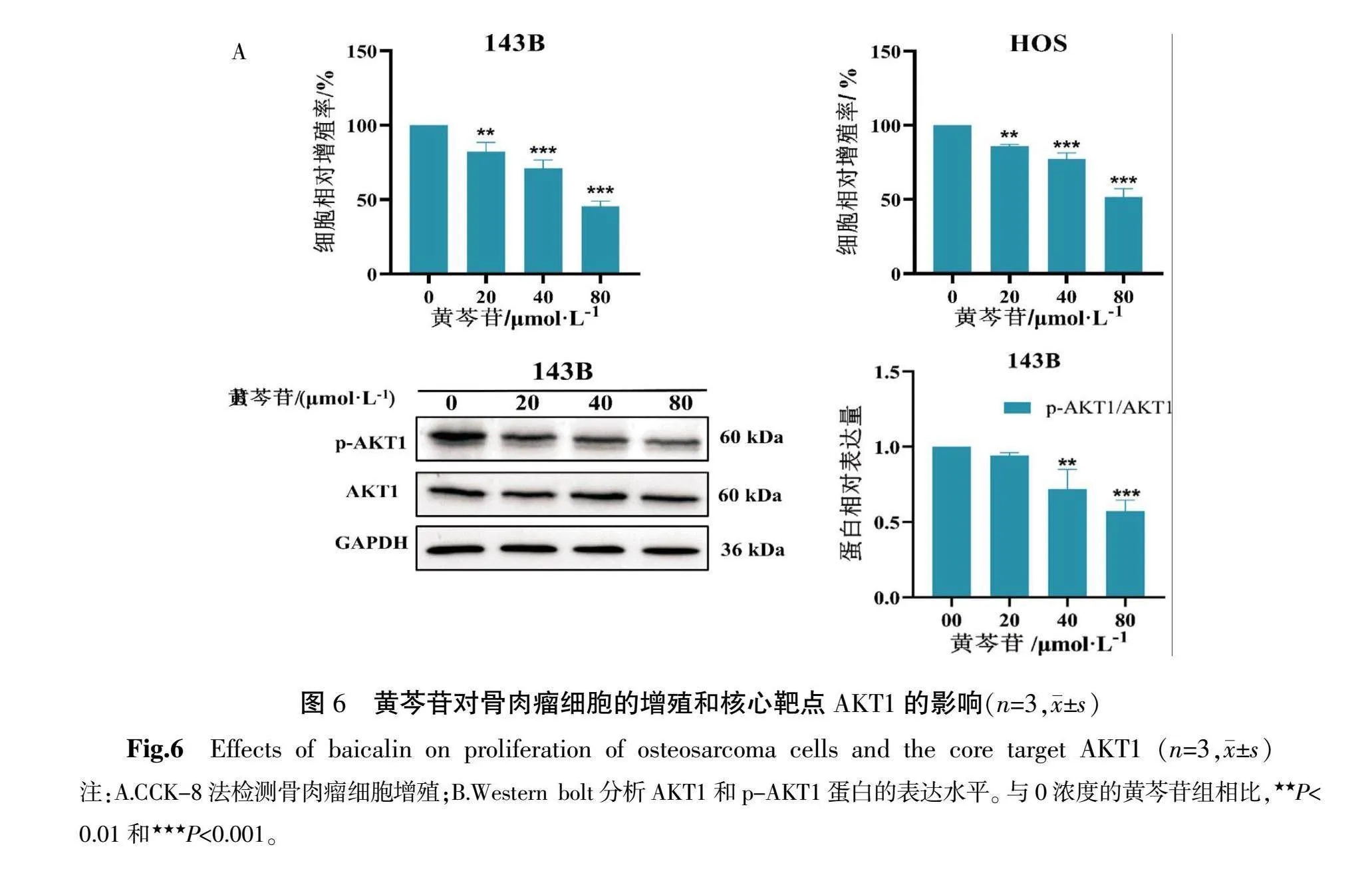
CCK-8细胞增殖结果显示,在0~80 μmol·L-1的黄芩苷浓度范围内,143B和HOS骨肉瘤细胞活力均明显降低(P<0.01)(图6A)。鉴于黄芩苷与核心靶点AKT1具有很好的结合活性,并且AKT1在PPI网络中的Degree值最高,进一步通过Western blot实验检测了黄芩苷对骨肉瘤细胞内AKT1蛋白水平变化的影响。与未处理的对照组相比,经黄芩苷处理后的143B细胞中AKT1蛋白的磷酸化水平显示出浓度依赖性的抑制效果(P<0.01)(图6B)。
4.7 SP600125增强黄芩苷抑制骨肉瘤细胞增殖和侵袭以及促进凋亡的作用
在研究SP600125单独或与黄芩苷联合对骨肉瘤细胞增殖、迁移和凋亡影响的过程中,CCK-8细胞增殖实验结果揭示,相较于单独使用黄芩苷,其与SP600125的联合应用能显著协同抑制143B和HOS两种骨肉瘤细胞系的增殖能力(P<0.05)。相比之下,SP600126仅在40 μmol·L-1浓度下才显示出对骨肉瘤细胞的抑制效应(P<0.01)(图7A)。在细胞迁移实验中,与空白组相比,单独应用黄芩苷可以增强对细胞迁移的抑制率(P<0.05)。而当SP600125与黄芩苷共处理时,这种联合作用可以进一步增强对细胞迁移能力的抑制效果(P<0.05)(图7B)。通过Annexin V-FITC/PI双染法评估细胞凋亡情况,流式细胞凋亡结果显示,与未处理的空白组相比,单独应用黄芩苷能增加细胞的凋亡率(P<0.01)。同时,SP600125与黄芩苷共处理时,可进一步增强黄芩苷诱导的细胞凋亡效果(P<0.05)(图7C)。此外,与空白组相比,黄芩苷组的凋亡相关Bcl-2蛋白表达量显著下降(P<0.01),而Bax蛋白表达量则显著增加(P<0.001)。当与SP600125联合处理时,相较于单独使用黄芩苷组,Bcl-2蛋白的表达量进一步降低(P<0.001),同时Bax蛋白的表达量也进一步增加(P<0.05)。这一结果进一步验证了SP600125和黄芩苷在促进细胞凋亡方面的协同增效作用(图7D)。
4.8 SP600125通过抑制AKT通路和调节自噬过程增强黄芩苷的抗骨肉瘤作用
KEGG富集分析的结果揭示了黄芩苷抗骨肉瘤作用与PI3K/AKT和MAPK信号通路的潜在关联。因此,本研究进一步检测了PI3K/AKT信号通路中AKT蛋白的表达变化。结果显示,黄芩苷的处理显著降低p-AKT/AKT的表达水平(P<0.001),并且联合SP600125共处理后,AKT磷酸化水平得到进一步抑制(P<0.05)(图8A)。为了进一步探讨JNK抑制剂SP600125如何增强黄芩苷诱导的细胞死亡效应。在处理143B细胞时,应用了80 μmol·L-1的黄芩苷或与SP600125的联合处理。透射电子显微镜结果显示,经黄芩苷处理的骨肉瘤细胞内自噬小体的数量显著增加。然而,联合SP600125时,由黄芩苷诱导的自噬小体的增多被明显减弱(图8B)。Western
blot结果显示,黄芩苷处理组的JNK蛋白被磷酸化激活。此外,自噬相关蛋白LC3-II与LC3-I的比值以及p62蛋白的表达均因黄芩苷的处理而显著增加(P<0.01)。然而,当使用SP600125抑制JNK时,黄芩苷所诱导的143B细胞中p62和LC3-II表达的激活被逆转(P<0.05)(图8C)。
5 讨论
本研究通过网络药理学共筛出46个共同靶点,其中,AKT1、CASP3、EGFR、HSP90AA1、ESR1和TNF等23个核心靶点与黄芩苷治疗骨肉瘤的机制密切相关。KEGG富集显示,PI3K-AKT信号通路、MAPK信号通路以及细胞凋亡等参与黄芩苷抗骨肉瘤的机制。核心靶点AKT1、HSP90AA1、ANXA5、CHEK1和PLAU在骨肉瘤组织中的表达显著高于正常骨组织。分子对接显示,黄芩苷与这些靶点都有较强的结合潜力。其中,AKT1与黄芩苷的结合具有很高的适配性,且在PPI网络中占据核心位置。另外,Western
blot结果显示,黄芩苷能降低143B细胞内AKT1蛋白的磷酸化水平,这为AKT1作为核心靶点提供了初步的验证。
骨肉瘤细胞的转移与预后不佳密切相关,是导致扩散、耐药及病情恶化的病理基础[13]。细胞凋亡机制是细胞受到特定信号的刺激,发生自主程序性细胞死亡,涉及染色质固缩、DNA断裂和凋亡小体形成的过程[14]。Bax与Bcl-2之间的比例变化能够影响线粒体膜电位,进而触发细胞色素C的释放,从而诱导Caspase系列反应,最终引发细胞凋亡[15]。目前,SP600125能协同增强抗肿瘤药物的治疗效果已在多种癌症模型中得到证实,与其他抗肿瘤药物联合使用来减弱肿瘤的耐药性[16]。本研究通过CCK-8测定和细胞迁移实验揭示,黄芩苷显著抑制骨肉瘤细胞的增殖和迁移能力。流式细胞凋亡检测和Western
bolt揭示了黄芩苷能明显诱导的骨肉瘤细胞的凋亡比例增高。此外,当SP600125与黄芩苷联合应用时,结果显示出SP600125能够协同增强黄芩苷抑制骨肉瘤细胞增殖和迁移的效果,并促进细胞凋亡。相比之下,单独使用JNK抑制剂SP600125对骨肉瘤细胞的影响却十分有限。
PI3K/AKT通路在骨肉瘤中被固定激活,并参与调控骨肉瘤细胞的转移、凋亡和自噬等过程[17]。此外,抑制该通路及其相关上下游分子的活性已成为骨肉瘤治疗的重要策略[18]。JNK是调控癌细胞凋亡和自噬的重要介质,它的激活在骨肉瘤细胞的生长、侵袭、凋亡和自噬等机制发挥重要作用[19-20]。研究表明,这两种信号通路存在相互干扰的现象,并组成一个复杂的调控网络[21]。有研究发现,SP600125通过增强柴胡皂苷D对AKT通路的阻断作用,抑制骨肉瘤细胞的恶性特性,并激活Caspase-3、Caspase-8和Caspase-9依赖的细胞凋亡[22]。自噬的激活通常与骨肉瘤细胞的增殖、转移、凋亡、化疗耐药和免疫治疗有关[23]。自噬与凋亡之间的相互作用构成了一个复杂且精确的平衡系统,其中一种过程的激活或抑制可以显著影响另一种过程的活性。此相互作用涉及众多信号通路及调节蛋白,包括Caspase-8、P53、Bcl-2和JNK等[24]。在自噬过程中,细胞质内LC3被水解成LC3-Ⅰ,与磷脂酰乙醇胺结合形成LC3-Ⅱ连接于自噬小体膜中[25]。此外,p62的表达与肿瘤形成、癌症促进和化疗耐药有关,是自噬的适配体[26]的LC3-Ⅱ和p62-Nrf2表达,活化凋亡相关蛋白的表达,触发自噬向凋亡的转换,增强C-2抗膀胱癌的敏感性[27]。此外,SP600125通过抑制JNK调控的自噬增强非小细胞肺癌细胞对mTORC1/2抑制剂的敏感性[28]。同时,在另一项研究中发现p62被pKAL以ROS依赖的方式显著上调,而SP600125抑制JNK介导p62的下调增强pKAL诱导的结直肠癌细胞凋亡[29]。本研究结果显示,黄芩苷和SP600125的联合应用能够有效抑制AKT的磷酸化,表明AKT通路参与其抗骨肉瘤的作用机制。透射电子显微镜观察显示,黄芩苷的处理能显著增加骨肉瘤细胞中自噬小体的数量。然而,当联用SP600125时,黄芩苷所诱导的自噬小体的增加受到抑制。另外,黄芩苷能增加自噬相关蛋白LC3-Ⅱ和p62的表达水平,以及促进JNK的磷酸化,而这种现象被JNK抑制剂SP600125所逆转。此外,SP600125的共处理增强了黄芩苷所诱导的细胞凋亡效应,这表明SP600125通过调节自噬过程,与黄芩苷协同发挥抗骨肉瘤的治疗效能。
综上所述,本研究运用了网络药理学和分子对接系统性分析黄芩苷抗骨肉瘤的潜在分子靶点和通路机制。在体外实验中,黄芩苷显著地抑制了骨肉瘤细胞的增殖与迁移,并有效地诱导骨肉瘤细胞凋亡。当与JNK抑制剂SP600125联合使用时,这一组合疗法通过降低AKT的磷酸化水平和调节自噬过程,进一步强化了对骨肉瘤治疗的协同效应。黄芩苷展现出针对多个靶点及其相应信号通路的显著作用,显示了其作为一种新型抗骨肉瘤药物的潜力。特别是与SP600125联合使用的策略,这为骨肉瘤的治疗提供了新的方向。
参考文献
[1] BIELACK S S, HECKER-NOLTING S, BLATTMANN C, et al. Advances in the management of osteosarcoma[J]. F1000Research, 2016, 5: 2767.
[2] BIELACK S, J?譈RGENS H, JUNDT G, et al. Osteosarcoma: The COSS experience[J]. Cancer Treatment and Research, 2009, 152: 289-308.
[3] YU X B, YUSTEIN J T, XU J M. Research models and mesenchymal/epithelial plasticity of osteosarcoma[J]. Cell & Bioscience, 2021, 11(1): 94.
[4] MEYERS P A, GORLICK R. Osteosarcoma[J]. Pediatric Clinics of North America, 1997, 44(4): 973-989.
[5] POSTHUMADEBOER J, WITLOX M A, KASPERS G J, et al. Molecular alterations as target for therapy in metastatic osteosarcoma: A review of literature[J]. Clinical & Experimental Metastasis, 2011, 28(5): 493-503.
[6] PRUDOWSKY Z D, YUSTEIN J T. Recent insights into therapy resistance in osteosarcoma[J]. Cancers, 2020, 13(1): 83.
[7] CZARNECKA A M, SYNORADZKI K, FIRLEJ W, et al. Molecular biology of osteosarcoma[J]. Cancers, 2020, 12(8): 2130.
[8] ZHANG Y, LOU Y N, WANG J B, et al. Research status and molecular mechanism of the traditional Chinese medicine and antitumor therapy combined strategy based on tumor microenvironment[J]. Frontiers in Immunology, 2020, 11: 609705.
[9] SINGH S, MEENA A, LUQMAN S. Baicalin mediated regulation of key signaling pathways in cancer[J]. Pharmacological Research, 2021, 164: 105387.
[10] WEN Y Q, WANG Y Z, ZHAO C X, et al. The pharmacological efficacy of baicalin in inflammatory diseases[J]. International Journal of Molecular Sciences, 2023, 24(11): 9317.
[11] ENNIS B W, FULTZ K E, SMITH K A, et al. Inhibition of tumor growth, angiogenesis, and tumor cell proliferation by a small molecule inhibitor of c-Jun N-terminal kinase[J]. The Journal of Pharmacology and Experimental Therapeutics, 2005, 313(1): 325-332.
[12] WU Q H, WU W D, JACEVIC V, et al. Selective inhibitors for JNK signalling: A potential targeted therapy in cancer[J]. Journal of Enzyme Inhibition and Medicinal Chemistry, 2020, 35(1): 574-583.
[13] CHONG Z X, YEAP S K, HO W Y. Unraveling the roles of miRNAs in regulating epithelial-to-mesenchymal transition (EMT) in osteosarcoma[J]. Pharmacological Research, 2021, 172: 105818.
[14] KOREN E, FUCHS Y. Modes of regulated cell death in cancer[J]. Cancer Discovery, 2021, 11(2): 245-265.
[15] CZABOTAR P E, GARCIA-SAEZ A J. Mechanisms of BCL-2 family proteins in mitochondrial apoptosis[J]. Nature Reviews Molecular Cell Biology, 2023, 24: 732-748.
[16] 莫艳秀, 彭凌音, 刘峻彤, 等. SP600125在肿瘤发生发展中的作用机制研究进展[J]. 湘南学院学报(医学版), 2022, 24(2): 68-73.
[17] ZHANG J, YU X H, YAN Y G, et al. PI3K/akt signaling in osteosarcoma[J]. Clinica Chimica Acta, 2015, 444: 182-192.
[18] KHEZRI M R, JAFARI R, YOUSEFI K, et al. The PI3K/AKT signaling pathway in cancer: Molecular mechanisms and possible therapeutic interventions[J]. Experimental and Molecular Pathology, 2022, 127: 104787.
[19] BUBICI C, PAPA S. JNK signalling in cancer: In need of new, smarter therapeutic targets[J]. British Journal of Pharmacology, 2014, 171(1): 24-37.
[20] 李国东, 蔡郑东, 张寅权, 等. 丝裂原活化蛋白激酶信号通路相关基因在人骨肉瘤中的表达[J]. 中华肿瘤杂志, 2009, 31(5): 340-345.
[21] ZHAO H F, WANG J, TONY TO S S. The phosphatidylinositol 3-kinase/akt and c-Jun N-terminal kinase signaling in cancer: Alliance or contradiction? (review)[J]. International Journal of Oncology, 2015, 47(2): 429-436.
[22] GAO T, ZHAO P, YU X L, et al. Use of Saikosaponin D and JNK inhibitor SP600125, alone or in combination, inhibits malignant properties of human osteosarcoma U2 cells[J]. American Journal of Translational Research, 2019, 11(4): 2070-2080.
[23] NING B, LIU Y X, HUANG T H, et al. Autophagy and its role in osteosarcoma[J]. Cancer Medicine, 2023, 12(5): 5676-5687.
[24] DAS S, SHUKLA N, SINGH S S, et al. Mechanism of interaction between autophagy and apoptosis in cancer[J]. Apoptosis, 2021, 26(9/10): 512-533.
[25] HECKMANN B L, GREEN D R. LC3-associated phagocytosis at a glance[J]. Journal of Cell Science, 2019, 132(5): jcs222984.
[26] KATSURAGI Y, ICHIMURA Y, KOMATSU M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor[J]. The FEBS Journal, 2015, 282(24): 4672-4678.
[27] YU H Y, WU C L, WANG X Y, et al. SP600125 enhances C-2-induced cell death by the switch from autophagy to apoptosis in bladder cancer cells[J]. Journal of Experimental & Clinical Cancer Research, 2019, 38(1): 448.
[28] JIN H O, HONG S E, PARK J A, et al. Inhibition of JNK-mediated autophagy enhances NSCLC cell sensitivity to mTORC1/2 inhibitors[J]. Scientific Reports, 2016, 6: 28945.
[29] JUNG E J, PARAMANANTHAM A, KIM H J, et al. Artemisia annua L. polyphenol-induced cell death is ROS-independently enhanced by inhibition of JNK in HCT116 colorectal cancer cells[J]. International Journal of Molecular Sciences, 2021, 22(3): 1366.