分子动力学模拟研究水/乙醇二元体系的热力学性质
2024-11-07周琳徐建强朱光来



摘要:采用分子动力学模拟方法研究了常温常压下水/乙醇体系的热力学性质,得到了不同浓度下二元体系的密度、自扩散系数、超额摩尔体积和径向分布函数。结果表明:随着乙醇摩尔分数的增加,二元体系的密度呈递减趋势,超额摩尔体积则先减小后增大且存在最小值,而乙醇的自扩散系数增大;结合径向分布函数分析可知,在乙醇低浓度时,水分子间的氢键网络结构较强,乙醇分子分散于其中,而随着乙醇浓度的增加,乙醇-乙醇分子之间的关联性增强,形成自己的氢键网络,而水分子仍倾向于自聚集,两种氢键网络相互交错。
关键词:分子动力学模拟;热力学性质;水/乙醇二元体系;径向分布函数
中图分类号:O552.4 文献标志码:A 文章编号:1001-2443(2024)04-0314-06
引言
氢键网络的存在使得水具有独特的性质,它不仅可以作为盐、蛋白质等多种分子的溶剂,还可以作为生物、化学反应过程的反应物[1-2]。研究水的结构及动力学性质, 对于认知其作为介质或反应物的作用机理有着重要的意义。根据与水的相互作用的不同,物质大致可分为疏水和亲水分子,分子的疏水性和亲水性的物理性质对于理解物质在液态水中的形态和溶解度至关重要。水的二元混合物往往会表现出非理想性,这通常与液体之间相互作用的性质和纳米级微观结构上的不均匀6U2Sc6cZx6oOlPXdngh82g==性有关[3]。但是,这些性质还没有被完全了解,近几十年以来,越来越多的研究聚焦于各种水溶液中水的性质与结构以及溶质的聚集现象[1-5]。
与水分子相比,乙醇等低级醇是同时具有甲基和羟基的有机小分子,结构上的差异使其具有两亲性特征[6, 7],因此成为分析更复杂的生物大分子行为的简单实验室模型:在醇的混合溶液中存在着氢键、疏水性和非极性等多种相互作用,这些是理解溶质分子在水溶液中的行为的关键。醇溶解于水中,会形成水-水、醇-醇以及水-醇复杂的氢键网络,使得混合物的结构和性质更加复杂[8-10],例如乙醇在水中形成胶束状结构,可能以五元环或六元环的形式存在[10]。醇水溶液,例如乙醇与水的二元混合溶液已广泛应用于从日常生活到科学研究的不同领域。理解液态醇-水体系中醇的聚集行为对于理解溶液体系中的溶解度和相分离等基本问题具有重要意义[1]。
近年来, 随着计算机技术的快速发展,分子动力学(MD)模拟已成为时下被广泛采用的计算庞大复杂系统的方法,它是建立系统的宏观行为与其分子间相互作用之间对应关系的强大技术[11]。采用分子动力学模拟技术可以提供系统的扩散性质与热力学性质等,在一些工业产品的过程开发和设计等方面正起着越来越重要的作用。通过调研发现,已有文献研究过乙醇混合物的部分性质,如扩散、径向分布函数、氢键分布等,但仍然有待进一步完善[12-15]。
本文基于SPCE水模型和OPLS-AA乙醇全原子力场,采用分子动力学模拟方法,对不同比例的水/乙醇二元体系进行系统研究,获得了二元体系的各项热力学性质。
1 力场模型与模拟方法
1.1 所用的力场模型和依据
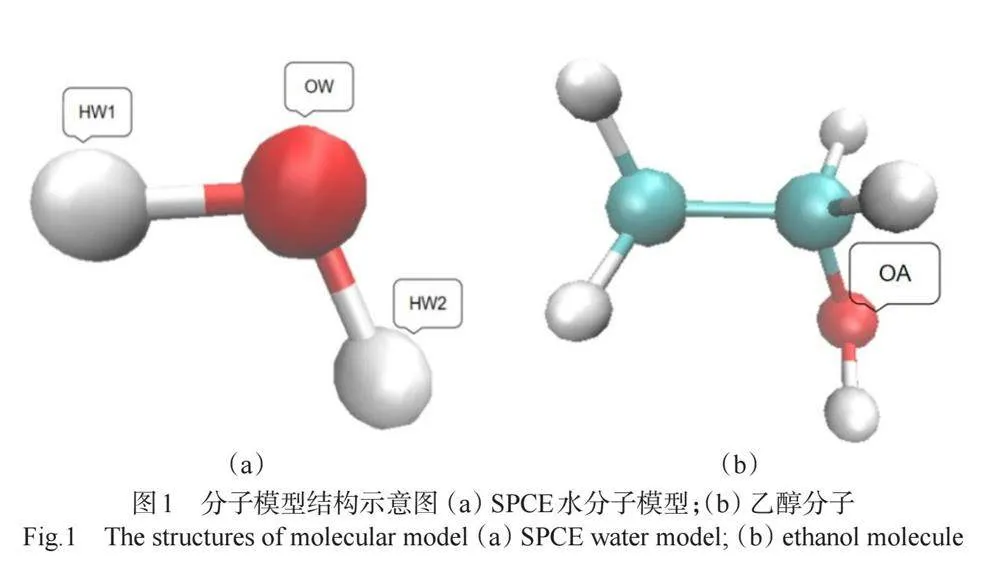
常用的水模型有SPC/SPCE、TIP3P、TIP4P等,我们在模拟过程中选择SPCE模型,如图1(a)所示。通过比较众多实验与模拟文献中的不同水模型的各种性质,我们可以看到SPC/SPCE水模型是一个形式简单又能反映体系本质的模型, 它反映了水的许多性质[6]。SPCE水模型是对SPC水模型的改进,虽然其密度值与实验值相比稍低,但是误差均在允许范围之内。而且从径向分布函数和凝结系数看[5],SPCE水模型得到的数值与实验结果吻合得更好,因此我们选用改进后的SPCE水模型。
乙醇分子采用OPLS-AA全原子模型[4],如图1(b)所示。乙醇模型可大致分为两类:以某一基团乙醇为单一相互作用位点的联合原子模型;将每个原子视为相互作用点的全原子模型。在适当的参数条件下,联合原子模型和全原子模型都能再现乙醇的性质。在模拟体系不太大的情况下,采用全原子模型能更加全面反映乙醇及其混合物的性质。
1.2 模拟细节
首先设置乙醇和水的比例,乙醇占混合溶液的摩尔分数[xi=NiN]设为0.0、0.2、0.4、0.5、0.6、0.8和1.0,其中Ni为乙醇的分子数、N是水加乙醇的分子总数。使用Packmol构建一个N为500的水和乙醇分子组成的体系,将其放在4×4×4 nm3的盒子里。然后运用Gromacs 2019.5[16]来进行MD模拟,在能量最小化之后,在NPT系综下运行20 ns,取最后5ns轨迹进行分析。模拟过程中温度保持在300 K以下,模拟步长设为2 fs,并且每2 ps取样一次。模拟过程中采用了周期性边界条件, 用V-rescale方法维持体系的温度,远程静电作用采用PME方法处理, 键长约束则采用LINCS算法。图2所示的是已平衡的二元体系(乙醇的摩尔分数为0.2)的快照图。
2 结果与讨论
2.1 密度
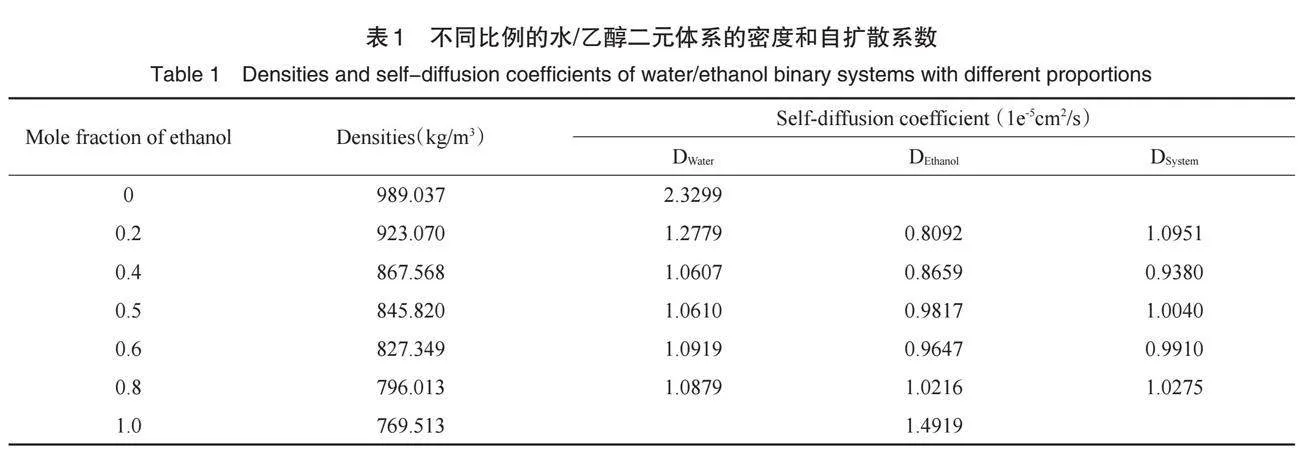
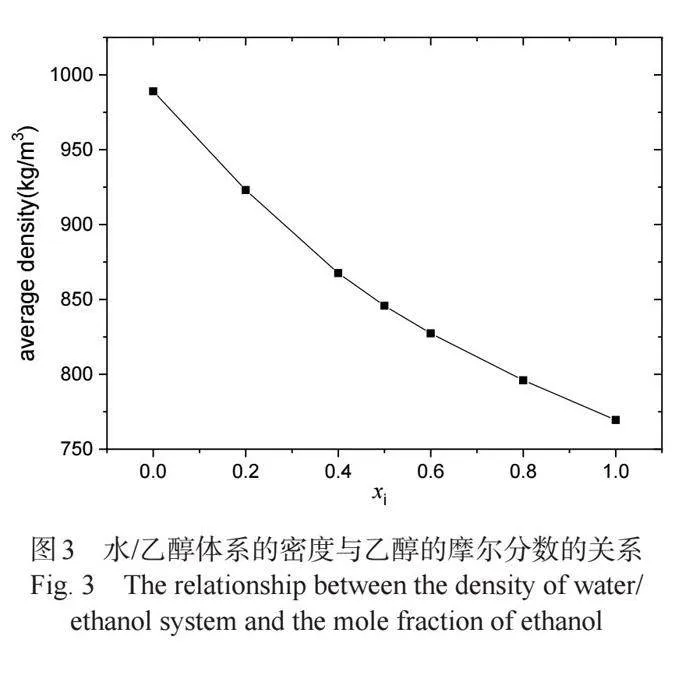
分析得到乙醇不同比例下的水/乙醇二元体系的密度列于表1中,并据此绘制了体系的平均密度曲线,如图3所示,其中纯水和乙醇的密度模拟值与实验值的误差均不超过2%,可见我们的模拟能较准确地预测体系的性质。从图中可以看到:乙醇浓度越高,体系的平均密度就越小,即对于混合体系,模拟得到的密度值随着乙醇浓度的增大逐渐减小,但并不呈线性变化规律,说明水与乙醇之间存在着复杂的相互作用,可能与体系中多种类型的氢键网络(水-水,乙醇-乙醇,水-乙醇)有关[1, 6]。
2.2 自扩散系数
MD模拟的优点之一在于不仅能获得平衡态性质,而且能获得动态性质,而自扩散系数作为人们在研究中最感兴趣的动态性质之一,其求法通常有两种:一是Green.Kubo法;二是Einstein法。本模拟采用的是Einstein法[14],如(1)式所示:
[D=16limt→∞ddt<[ri(t)-ri(0)]2>] (1)
其中D为扩散系数,t为时间,ri(t)为t时刻i组分质心的位置,ri(0)为0时刻i粒子的位置。其几何解释是扩散系数为均方位移(Mean Square Displacement,简称MSD)对时间作图趋于无穷时斜率的l/6。
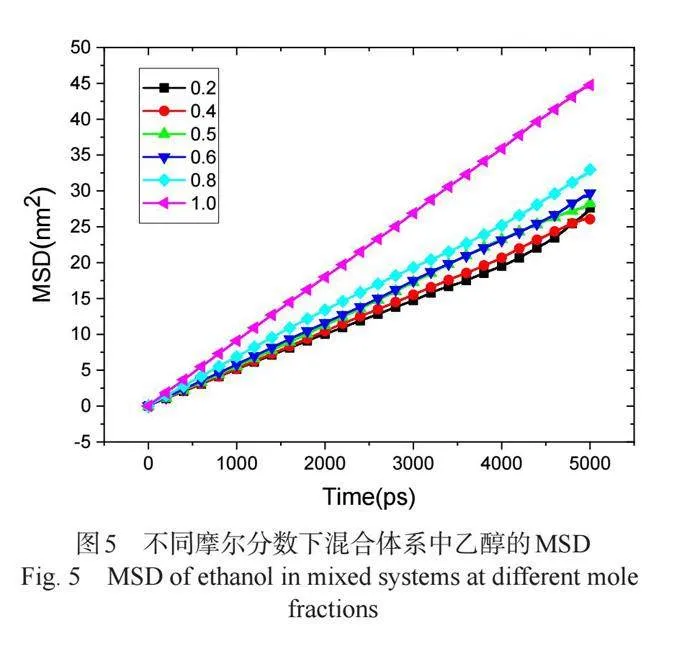
选择乙醇摩尔分数为0.2时的体系,观察乙醇、水和系统的MSD曲线,如图4所示,可以发现系统的MSD曲线差不多是水和乙醇的MSD曲线的平均。图5所示是不同比例下乙醇的MSD曲线,从图中可知,随着乙醇浓度的增加,乙醇的MSD曲线的斜率逐渐增大。这一现象与文献中的结论类似[10],表明模拟结果是合理性的。
通过对MSD曲线进行拟合可以得到各组分的自扩散系数,相关结果列在表1中。通过对比表1中的数据并结合文献分析可以发现[12],二元体系的总自扩散系数与浓度相关度不大,基本维持在1×10-5 cm2·s-1左右;而随着乙醇浓度的增加,乙醇的自扩散系数总体是增加的,而水的自扩散系数在一开始快速下降后保持一定水平几乎不变。说明水和乙醇之间存在比较强的相互作用,且这种作用可能不仅仅只限于分离的分子间作用力,在乙醇浓度较高的情况下,水分子原本的网状氢键结构仍然得以保持,并可能与乙醇分子形成的氢键相互结合,这点也可以从后续对相关原子间的径向分布函数分析得到进一步证实。
除此以外,通过表1还可以发现,在乙醇摩尔分数为0.2时,乙醇的自扩散系数最小;而在乙醇摩尔分数为0.4时,水的自扩散系数最小,两者变化并不同步,这与乙醇和水的竞争性自缔合有关,即使在乙醇浓度非常高的情况下,水-水间的氢键仍然会存在[9],它们的自扩散系数与乙醇和水的聚集体的运动相关联。
2.3 超额摩尔体积
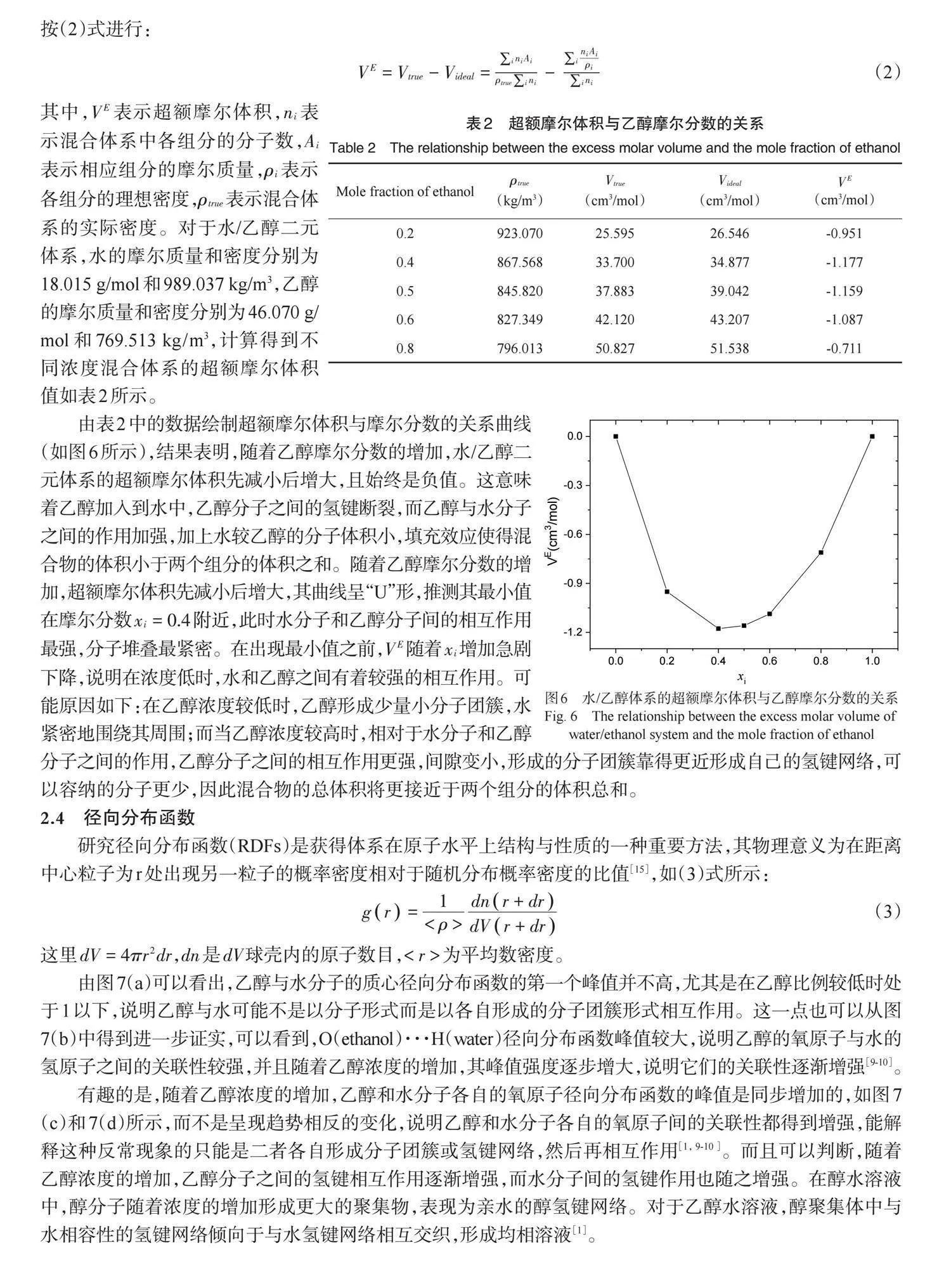
为进一步分析水分子与乙醇分子之间的相互作用,我们分析了二元体系的超额摩尔体积[6],具体计算可按(2)式进行:
[VE=Vtrue-Videal=iniAiρtrueini-iniAiρiini] (2)
其中,[VE]表示超额摩尔体积,[ni]表示混合体系中各组分的分子数,[Ai]表示相应组分的摩尔质量,[ρi]表示各组分的理想密度,[ρtrue]表示混合体系的实际密度。对于水/乙醇二元体系,水的摩尔质量和密度分别为18.015 g/mol和989.037 kg/m3,乙醇的摩尔质量和密度分别为46.070 g/mol和769.513 kg/m3,计算得到不同浓度混合体系的超额摩尔体积值如表2所示。
由表2中的数据绘制超额摩尔体积与摩尔分数的关系曲线(如图6所示),结果表明,随着乙醇摩尔分数的增加,水/乙醇二元体系的超额摩尔体积先减小后增大,且始终是负值。这意味着乙醇加入到水中,乙醇分子之间的氢键断裂,而乙醇与水分子之间的作用加强,加上水较乙醇的分子体积小,填充效应使得混合物的体积小于两个组分的体积之和。随着乙醇摩尔分数的增加,超额摩尔体积先减小后增大,其曲线呈“U”形,推测其最小值在摩尔分数[xi=0.4]附近,此时水分子和乙醇分子间的相互作用最强,分子堆叠最紧密。在出现最小值之前,[VE]随着[xi]增加急剧下降,说明在浓度低时,水和乙醇之间有着较强的相互作用。可能原因如下:在乙醇浓度较低时,乙醇形成少量小分子团簇,水紧密地围绕其周围;而当乙醇浓度较高时,相对于水分子和乙醇分子之间的作用,乙醇分子之间的相互作用更强,间隙变小,形成的分子团簇靠得更近形成自己的氢键网络,可以容纳的分子更少,因此混合物的总体积将更接近于两个组分的体积总和。
2.4 径向分布函数
研究径向分布函数(RDFs)是获得体系在原子水平上结构与性质的一种重要方法,其物理意义为在距离中心粒子为r处出现另一粒子的概率密度相对于随机分布概率密度的比值[15],如(3)式所示:
[gr=1<ρ>dnr+drdVr+dr] (3)
这里[dV=4πr2dr],[dn]是[dV]球壳内的原子数目,[<r>]为平均数密度。
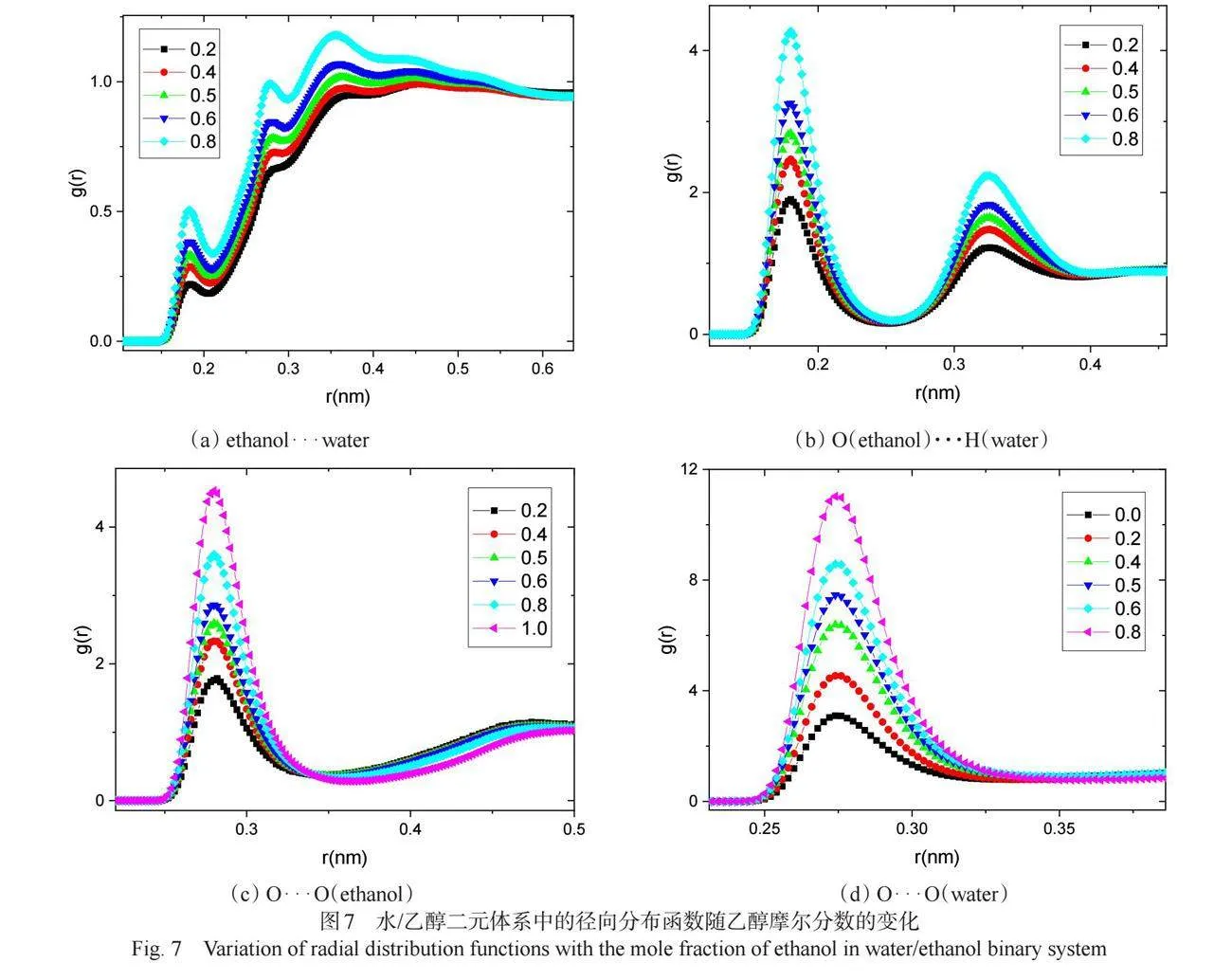
由图7(a)可以看出,乙醇与水分子的质心径向分布函数的第一个峰值并不高,尤其是在乙醇比例较低时处于1以下,说明乙醇与水可能不是以分子形式而是以各自形成的分子团簇形式相互作用。这一点也可以从图7(b)中得到进一步证实,可以看到,O(ethanol)···H(water)径向分布函数峰值较大,说明乙醇的氧原子与水的氢原子之间的关联性较强,并且随着乙醇浓度的增加,其峰值强度逐步增大,说明它们的关联性逐渐增强[9-10]。
有趣的是,随着乙醇浓度的增加,乙醇和水分子各自的氧原子径向分布函数的峰值是同步增加的,如图7(c)和7(d)所示,而不是呈现趋势相反的变化,说明乙醇和水分子各自的氧原子间的关联性都得到增强,能解释这种反常现象的只能是二者各自形成分子团簇或氢键网络,然后再相互作用[1, 9-10 ]。而且可以判断,随着乙醇浓度的增加,乙醇分子之间的氢键相互作用逐渐增强,而水分子间的氢键作用也随之增强。在醇水溶液中,醇分子随着浓度的增加形成更大的聚集物,表现为亲水的醇氢键网络。对于乙醇水溶液,醇聚集体中与水相容性的氢键网络倾向于与水氢键网络相互交织,形成均相溶液[1]。
综合对图7的分析,可以发现所列出的原子径向分布函数的第一个峰值均表现显著, 反映出它们的分布具有近程有序性。随着乙醇浓度的增加,乙醇和水分子间的关联性增强,乙醇-乙醇、水-水的氢键也逐渐增强。由文献可知[9],在乙醇浓度较低的溶液中,水分子间的氢键结构较强,乙醇-乙醇的氢键结构逐渐断裂,乙醇分子分散在水分子的氢键网络中;随着乙醇浓度的增加,乙醇-乙醇分子之间的关联性增强,形成自己的氢键网络,而水分子也倾向于自我聚集,所以水分子间的关联性并没有由于水分子数量减少而减弱。乙醇在高浓度溶液中的聚集物形成了一个扩展的与水相容的氢键网络,且O(ethanol)···H(water)之间的相互作用由于较强氢键而逐渐增强[1],两种的氢键网络之间的相互作用导致了醇-水关联性的增强。
3 结论
本文采用SPCE水模型和OPLS全原子乙醇分子模型,对比分析了水/乙醇二元体系的密度、自扩散系数、超额体积、径向分布函数等性质。主要结论如下:随着乙醇浓度的增加,体系的密度非线性减小,乙醇的自扩散系数增大但水并不同步变化,体系的超额摩尔体积则先减小后增大。在低乙醇浓度时,主要是乙醇分子与水的氢键网络间的相互作用;而随着乙醇浓度的增加,乙醇-乙醇的氢键结构开始形成并逐步增强,水-乙醇的关联性也增强,水和乙醇的氢键网络结构相互交错;但即使在乙醇浓度非常高的情况下,水分子之间的氢键作用仍存在,且水分子间的关联和纯水相比反而明显加强。我们的工作进一步揭示了水/乙醇二元体系中复杂的相互作用,对理解醇/水体系中的聚集和相行为具有一定参考价值。
参考文献:
[1] CHOI S, PARAMESWARAn S, CHOI J H. Understanding alcohol aggregates and the water hydrogen bond network towards miscibility in alcohol solutions: graph theoretical analysis[J]. Phys Chem Chem Phys, 2020, 22: 17181-17195.
[2] 周健, 陆小华, 王延儒, 等. 液体水的分子动力学模拟[J]. 南京化工大学学报, 1998, 20(3): 1-5.
[3] ESSAFRI I, GHOUFI A. Microstructure of nonideal methanol binary liquid mixtures[J]. Phys Rev E, 2019, 99(6): 062607.
[4] JORGENSON W L, CHANDRASEKHAR J, MADURA J D, et al. Comparison of simple potential functions for simulating liquid water [J]. J Chem Phys, 1983, 79(2): 926-930.
[5] 李印实, 何雅玲, 孙杰, 等. 不同水分子模型凝结系数的分子动力学模拟对比研究[J]. 西安交通大学学报, 2006, 40(11): 1272-1275.
[6] PÉREZ EG, GONZÁLEZ-SALGADO D, Lomba E. Molecular dynamics simulations of aqueous solutions of short chain alcohols. excess properties and the temperature of maximum density[J]. Fluid Phase Equilibr, 2021, 528: 112840.
[7] 曾勇平, 朱晓敏, 杨正华. 水、甲醇和乙醇液体微结构性质的Car-Parrinello分子动力学模拟[J]. 物理化学学报, 2011, 27(12): 2779-2785.
[8] 马宗平, 唐耀, 刘娟红, 等. 外磁场作用下乙醇团簇的分子动力学模拟[J]. 西南民族大学学报(自然科学版), 2009, 35(5): 1053-1056.
[9] 张翠娟, 程岳山. 分子动力学模拟乙醇/水二元混合物的扩散性质[J]. 泰山学院学报, 2011, 33(6): 86-91.
[10] GEREBEN O. Ring structure analysis of ethanol-water mixtures[J]. J Mol Liq, 2015, 211: 812-820.
[11] 陈正隆, 徐为人, 汤立达. 分子模拟的理论与实践[M]. 北京:化学工业出版社, 2007: 1-3.
[12] 田国才, 王丁, 王鹏飞. 离子液体[EMIM][BF4]/乙醇混合体系的分子动力学模拟研究[J]. 昆明理工大学学报(自然科学版), 2012, 37(6): 21-27.
[13] NOSKOV S Y, Lamoureux G, Roux B. Molecular dynamics study of hydration in ethanol-water mixtures using a polarizable force field[J]. J Phys Chem B, 2005, 109: 6705-6713.
[14] ZHANG C J, YANG X N. Molecular dynamics simulation of ethanol/water mixtures for structure and diffusion properties[J]. Fluid Phase Equilibr, 2005, 231: 1-10.
[15] MIJAKOVIĆ M, KEŽIĆ B, ZORANIĆ L, et al. Ethanol-water mixtures: Ultrasonics, Brillouin scattering and molecular dynamics[J]. J Mol Liq, 2011, 164: 66-73.
[16] VAN DER SPOEL D, LINDAHL E, HESS B, et al. GROMACS: fast, flexible and free[J]. J Comp Chem, 2005, 26(16): 1701-1718.
Thermodynamic Properties of Water/Ethanol Binary System Studied by Molecular Dynamics Simulation
ZHOU Lin, XU Jian-qiang, ZHU Guang-lai
(Anhui Province Key Laboratory of Optoelectronic Materials Science and Technology, School of Physics and Electronic Information, Anhui Normal University, Wuhu 241002, China)
Abstract: The thermodynamic properties of water/ethanol binary system at normal temperature and pressure were studied by molecular dynamics simulation. The density, self-diffusion coefficient, excess molar volume and radial distribution function of mixed system at different concentration were obtained. The results show that with the increase of the mole fraction of ethanol, the density of the mixed system decreases, the excess molar volume first decreases and then increases with a minimum value, and the self-diffusion coefficient of ethanol increases. Combined with the analysis of radial distribution function, it is known that when the concentration of ethanol is low, the hydrogen bonding network structure among water molecules is strong, and ethanol molecules are dispersed in it. With the increase of ethanol concentration, the correlation among ethanol molecules increases and forms their own hydrogen bonding network, while water molecules still tend to gather themselves, and the two hydrogen bonding networks are interlaced with each other.
Key words: molecular dynamics simulation; thermodynamic properties; water/ethanol binary system; radial distribution functions
(责任编辑:马乃玉)