基于门控单元的农作物蛋白质磷酸化预测模型研究
2024-08-06段旭福李重



关键词:深度学习;生物信息学;蛋白质磷酸化;计算生物学
中图分类号:TP389.1 文献标志码:A
0 引言(Introduction)
近年来,植物病理学的深入研究,极大地增进了我们对植物与病原菌相互作用机制的认知,也揭示了蛋白质磷酸化在诸多生物学过程中的关键作用,为农作物病害的防控提供了新的视角[1]。对蛋白质磷酸化的迅速判断,对农作物病害的有效防治也变得愈发重要。
然而,传统的蛋白质磷酸化的检测分析多采用实验方法,如液相色谱串联质谱、放射性化学标记和免疫检测、邻近连接分析、染色质免疫沉淀和蛋白质印迹[2]。这些技术通常耗时且劳动强度大。整个实验过程耗时较长,并且需要实验人员具备较高的专业技能和经验,这在一定程度上限制了蛋白质磷酸化研究的规模和效率。随着技术的进步和新计算方法的涌现,研究人员开发了许多基于智能算法的工具,这些工具极大地丰富了该领域的研究手段[3]。但是,目前的计算方法通常无法同时满足使用简单、快速检测、高精度等需求,本研究致力于开发一种高效、精准且操作简便的计算方法,旨在实现磷酸化位点的快速检测。
1 相关理论(Related theory)
1.1 蛋白质磷酸化
蛋白质磷酸化是一种生物学过程,其中磷酸基团被共价地添加到蛋白质分子的特定氨基酸残基上。这一修饰过程通常通过激酶酶类催化,其在细胞内发挥着关键的调控作用。磷酸基团的添加可以改变蛋白质的结构、功能和相互作用机制,影响其在细胞内的活性和稳定性[4]。通常,酪氨酸(Tyr)、丝氨酸(Ser)和苏氨酸(Thr)是常见的磷酸化位点,在这些磷酸化位点上,氨基酸的OH 基团与ATP的γ-磷酸基团形成磷酸酯,而这些位点是磷酸化研究中受到广泛研究的对象。
1.2 磷酸化位点预测
在蛋白质工程领域,深度学习的应用日益凸显其重要性,它依托于蛋白质序列和结构等丰富数据作为输入,通过生成特征并采用不同的算法进行模型构建和优化。这一方法为更精确地分类和寻找磷酸化位点提供了新的途径。随着生物大数据集的构建和计算能力的提升,越来越多的计算方法被提出并用于磷酸化位点的预测。KHALILI等[5]使用处理表格数据的深度学习模型训练了一个大豆蛋白磷酸化预测器。LV等[6]使用卷积神经网络-长短期记忆网络(CNN-LSTM)识别感染SARS冠状病毒2型(SARS-CoV-2)的宿主细胞中的磷酸化位点。WANG等[7]提出了一个名为TransPhos的预测器,用于预测磷酸化位点。这些研究表明,深度学习方法在磷酸化位点预测方面取得了显著的进展。
1.3 门控机制
门控机制在神经网络中的应用由来已久,常使用Sigmod 函数或Tanh函数控制信息流的比例,是一个决定特征是否继续流入下一层的控制器。gMLP[8]是一种基于MLP与门控机制的简单的神经网络架构,它融合了线性空间投影和乘法门控机制,在掩码语言建模方面取得了出色的结果,甚至在参数更少的情况下其性能超过了一些基于Transformer的模型。在增加数据和计算能力的情况下,具有类似gMLP这样简单的空间交互机制的模型,已经展现出了与Transformer相媲美的强大性能。gMLP主要依靠静态参数化的通道映射(channelprojections)和空间映射(spatial projections),由L 个相同结构和大小的模块组成,X∈ n×d 代表长度为n 且序列维度是d 的向量表示,每个模块可以表示为
2 方法概述(Methodology overview)
2.1 数据预处理
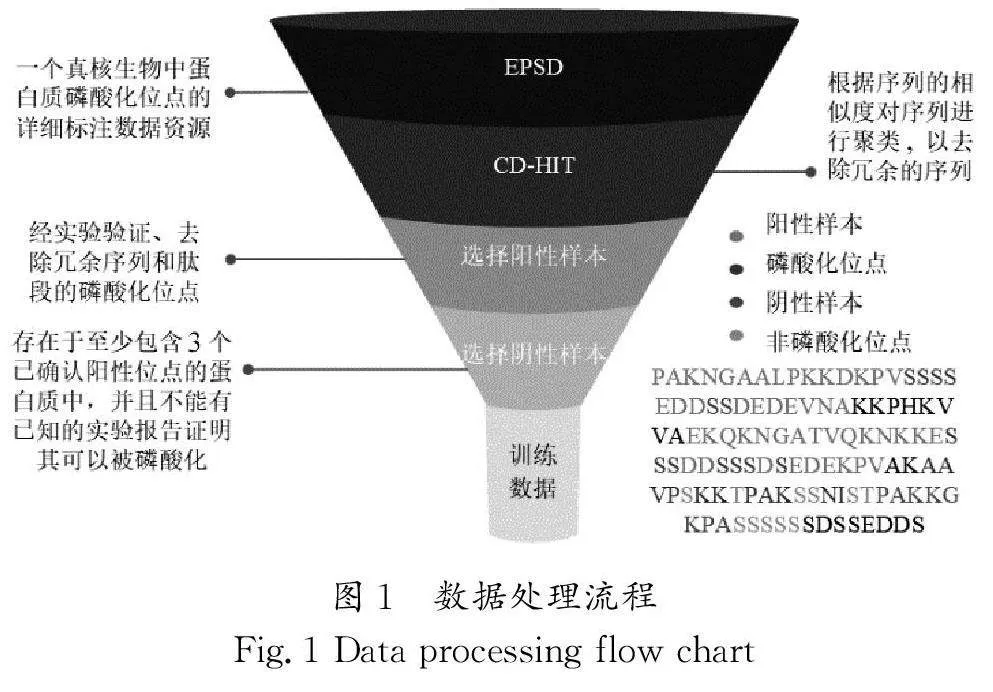
在以往的研究中,通常遵循3个原则进行数据预处理:①作为阳性样本的磷酸化位点经过实验验证;②使用聚类工具删除序列相似性过高的蛋白质序列;③随机选取的阴性位点,完整的蛋白质序列中至少有3个已确认阳性位点。
真核生物磷酸化位点数据库(Eukaryotic PhosphorylationkJR8M4CmiapE6wNXAznypoTAg0zeBausBnu8vZfjJQo=Site Database,EPSD)由LIN等[9]重新整理,是近期更新的最具体和最全面的磷酸化位点数据库之一,本研究采用该数据库作为主要的数据来源,并延续以往的经验,采用相似的数据处理过程。为避免阳性和阴性数据集中存在同源序列导致预测模型的性能被高估,利用CD-HIT(一种用于聚类相似生物序列的工具)[10]以40%的序列相似性为标准,对磷酸化蛋白质序列进行聚类,具有中心为丝氨酸、苏氨酸和酪氨酸残基及经实验验证的磷酸基团的肽链,被视为阳性样本,并选择15作为采样窗口大小(在选定的磷酸化位点上左、右各取15个氨基酸组成肽链),随机选择一部分与阳性样本数量相当的非冗余的阴性样本,以平衡数据集。
根据上述条件进行样本提取时,可能存在大量的经实验验证的磷酸化位点在肽链中排列过于紧密,导致同一个肽段反复被添加到阳性样本中,本研究采用一种新的采样方式,即在同一个采样窗口内仅采样一次,跳过那些在同一个窗口中过于密集的肽段。在第一个采样窗口中,选中一个磷酸化位点后,右边的窗口中符合要求的阳性样本将不再被考虑,阴性样本同理,同时选取阴性样本时,还要考虑不与阳性样本的窗口重叠。图1为数据处理流程。
2.2 序列特征
实验中涉及的氨基酸包括构成生物体的20种标准氨基酸和由基因密码子直接编码的2种非标准氨基酸,以数字1~22 对其进行编码。在蛋白质研究中,为了方便计算机处理和分析,研究者通常将不同类型的氨基酸以数字形式进行编码。这种编码方式的选择是为了将具有不同性质的氨基酸转化为统一的数字表示,从而将目标肽链转化为L×1的向量(L 表示肽链的长度)。每一个数字都代表特定的氨基酸类型。
2.3 蛋白质内在无序性得分
近年来的研究表明,蛋白质中存在一些并没有固定结构的无序区域,这些区域在许多细胞过程中发挥着重要的功能作用,并且与蛋白质之间的相互作用密切相关[11]。本研究采用IUPred3(Intrinsically Unstructured Protein Predictor)[12]工具获取蛋白质内在无序性得分,它依赖于能量估计方法,能预测每个氨基酸处于无序区域的趋势。对于长度为m 的氨基酸序列S,构建一个m×3的向量,分别代表短无序评分(缺乏稳定的三维结构且长度不超过30个残基的肽段)和长无序评分(长度超过30个残基的肽段)及ANCHOR(Analyzing the Chainof Ordered Regions)分数。
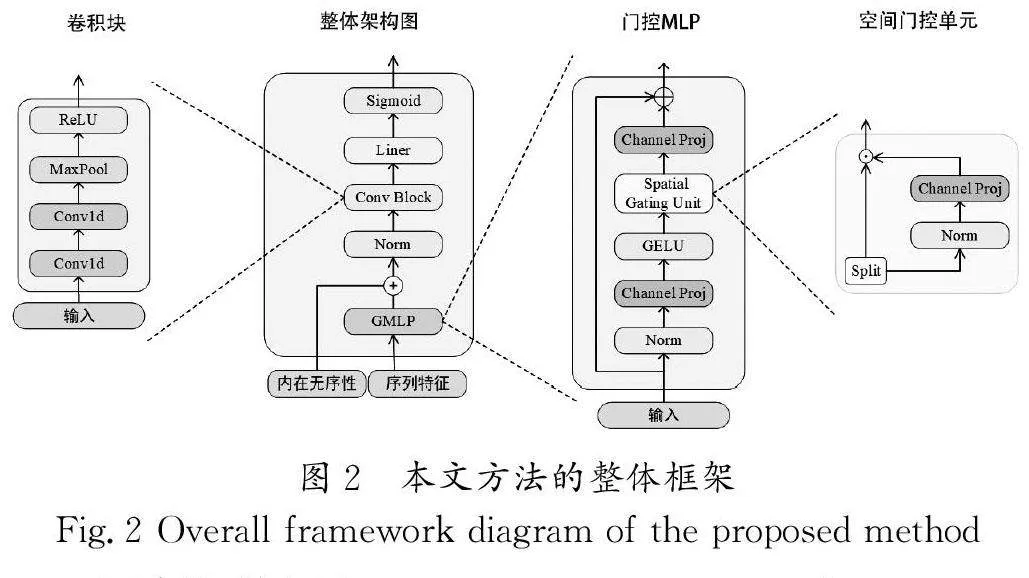
2.4 方法整体架构
本研究构建了一个网络架构,以gMLP作为编码层。将蛋白质转为数据特征后经过gMLP编码,得到的语义信息将与内在无序性得分进行拼接。为了提升模型的表达能力,对数据进行了标准化。对数据依次进行卷积、池化等操作,其中卷积操作有助于提取蛋白质的语义特征,而池化操作则可以减小特征图的尺寸。
为了将提取的特征有效地映射到最终的输出空间,引入了线性层。采用Sigmoid激活函数将输出映射到0~1,以便进行二分类。这样的设计不仅能有效地处理蛋白质序列的语义信息,还能充分利用内在无序性得分,为蛋白质研究和分类任务提供更为有效的工具。图2为本文方法的整体框架。
3 实验和结果(Experiment and result)
3.1 数据集划分
本研究按照64%、16%、20%的比例将数据划分为训练集、验证集和测试集(按照五倍交叉验证的原则,即1∶4的比例划分测试集和训练集,再将训练集的20%划为验证集),数据划分如图3所示,并在此基础上进行模型的训练和评估。
在模型训练的过程中,引入学习率调度器,在训练的不同阶段动态地调整学习率,以更好地适应数据分布的变化。将初始学习率设置为0.001,并设定了每隔10个批次,学习率以0.9的比例进行衰减。采用这一学习率调整策略旨在训练初期使用较大的学习率使模型更快收敛,随着训练的进行,逐渐减小学习率,有助于模型更精细地学习数据的特征。
3.3 评价指标
本研究中所用评价指标包括准确率(Accuracy,ACC)、AUC-ROC曲线下面积(Area Under the Curve,AUC)、特异性(Specificity,SP)、精确率(Precision,PRE)、召回率(Recall)、F1 分数(F1 Score,F1)和马修斯相关系数(Matthews CorrelationCoefficient,MCC)。
ACC 是分类模型正确预测的样本数占总样本数的比例,表示正确分类样本占总样本数的百分比;AUC 是AUC-ROC 曲线下的面积,用于度量二分类模型的性能,范围为0~1,数值越大,表示模型性能越好;Recall(真正例率)是真正例在实际正例中的比例,表示在所有实际正例中,模型正确预测为正例的比例;SP(真负例率)是真负例在实际负例中的比例,表示在所有实际负例中,模型正确预测为负例的比例;PRE(真正例率)表示在模型预测为正例的样本中,实际为正例的比例;F1 是精确率和召回率的调和平均值,用于综合考虑分类模型的性能衡量模型在精确率和召回率之间的平衡;MCC是衡量二分类模型性能的综合指标,常用于衡量模型的综合性能,尤其在不平衡数据集中更具优势。
3.4 在不同物种数据集上的性能比较
针对不同的农作物品种蛋白质,采用相同的数据处理方式分别进行训练。所用的数据均从EPSD[9]数据库中获取,以小麦、水稻亚种-粳稻、水稻亚种-籼稻、玉米及大豆为例,表1中展示了使用本文模型训练上述数据集在五倍交叉验证下的准确率、AUC-ROC曲线下面积、特异性、精确率、召回率、F1分数及马修斯相关系数。正、负样本的筛选与比例,以及训练集、验证集和测试集的划分均按照前文描述的方法进行。各农作物所使用的训练样本数量详见表2。
3.5 与其他方法的比较
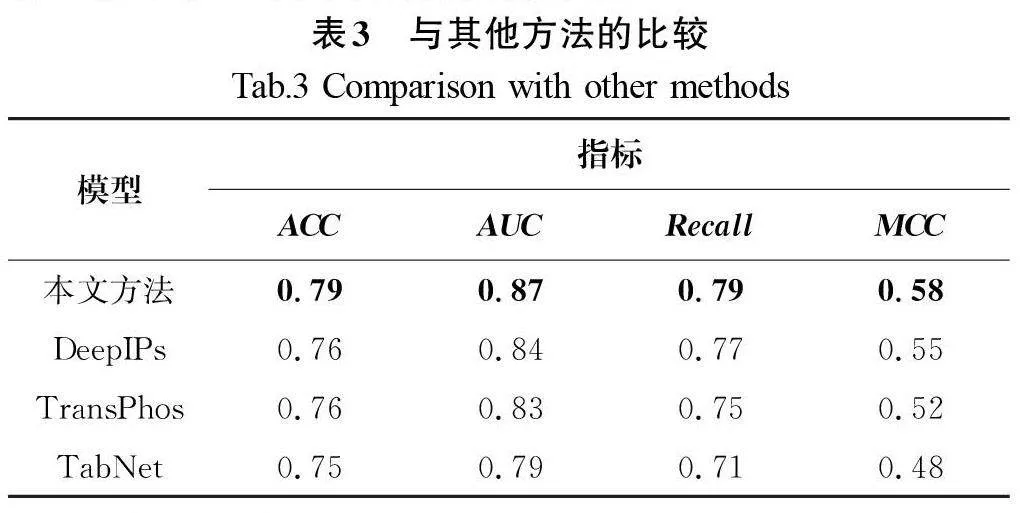
为了评估本研究提出模型的预测能力,将其与另外3种方法进行了比较,包括DeepIPs、TabNet和TransPhos,并采用五倍交叉验证进行了验证。
DeepIPs[6]是一个专门用于识别SARS-CoV-2感染宿主细胞中磷酸化位点的深度学习模型,通过词嵌入方法和CNNLSTM架构进行特征提取和分类。
TabNet模型由ARIK等[13]提出,主要用于表格数据集,KHALILI等[5]首次将其用于处理和分析生物数据,并取得了良好的效果。
TransPhos[7]是一个专门用于预测蛋白质磷酸化位点的深度学习模型,由基于Transformer编码器和密集连接的卷积神经网络块构成。
以玉米磷酸化位点的预测为例,本研究使用相同的数据进行5种方法的训练。正、负样本的总数分别为7 729个,其中Ser/S、Thr/T和Tyr/Y的数量分别为12 244个、2 724个、490个。使用相同的随机种子,表3展示了使用不同方法训练玉米数据集在五倍交叉验证下的部分关键指标。
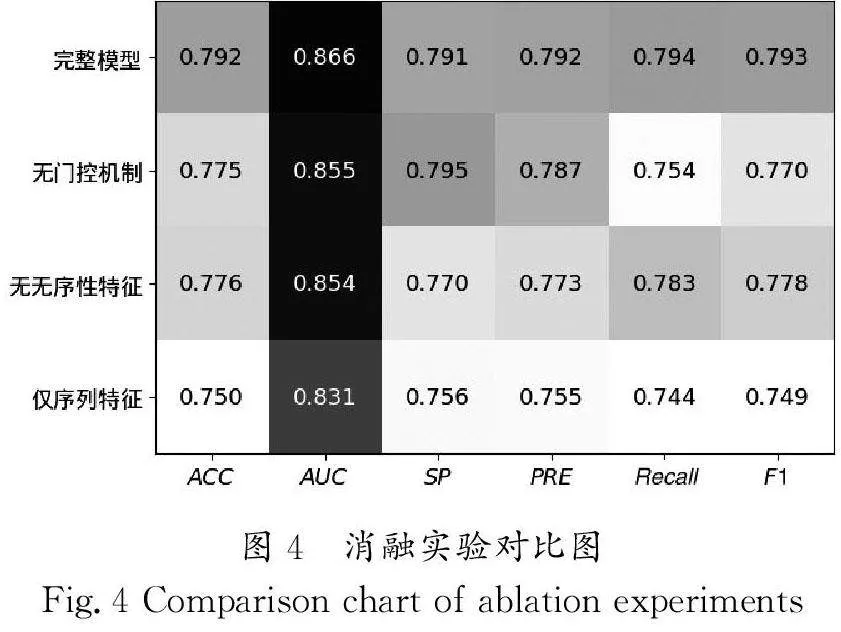
3.6 消融实验
为探索该模型不同部分的贡献,本研究进行了一系列实验,评估了该方法在缺失不同内容时对整体性能的影响。本研究对使用gMLP与否和使用无序性得分作为特征与否进行了组合验证。图4中为五倍交叉验证中的ACC、AUC、SP、PRE、Recall、F1分数的平均值。这些实验结果也验证了在模型中引入内在无序性得分和gMLP的有效性,并为其在实际应用中的可靠性提供了有力支持。
4 结论(Conclusion)
在本研究提出的方法中,使用gMLP作为特征提取器,引入门控机制,更高效地利用了蛋白质语义信息流;同时,优化了数据采样方式,每个窗口内仅采样一次,避免了同源肽段被频繁添加至训练集;此外,引入内在无序性得分作为特征,使模型能够学习到更多的蛋白质语义关联信息。实验结果表明,该方法能够有效提升预测精度,并且优于基于Transformer模型的方法,仅使用从序列中提取的特征,避免了复杂的特征提取操作,显著降低了计算成本,对计算资源没有较高的要求且操作简单。该方法相较于目前先进的计算方法(如DeepIPs、TabNet、TransPhos)在磷酸化位点预测上的表现更为出色,为农作物病害的深入研究和治理提供了一种更为高效和可行的途径。
作者简介:
段旭福(1998-),男,硕士生。研究领域:深度学习,蛋白质组学。
李重(1975-),男,博士,教授。研究领域:计算生物学,人工智能与数据分析,图形图像与虚拟现实。
