一种二进制癌症单驱动通路识别模型和算法
2024-07-31张奕鲁贺
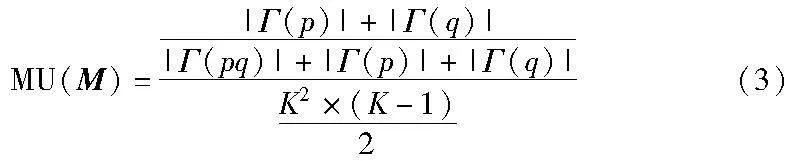



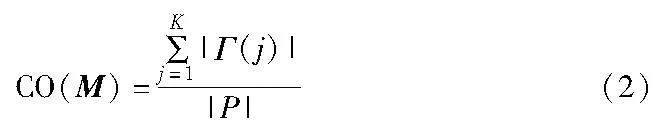
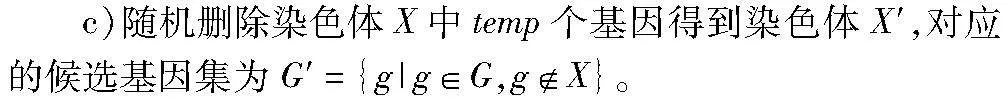
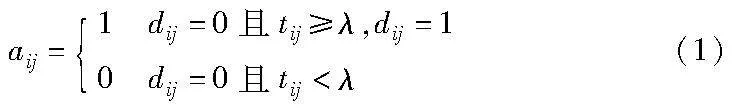
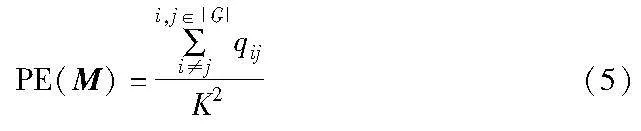

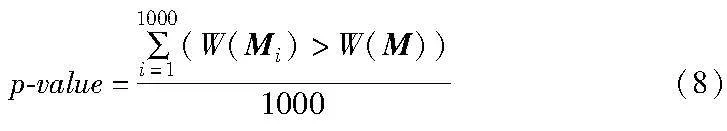
摘 要:针对驱动通路识别的相关研究依赖传统生物实验方法,存在费时费力且经济成本高的问题,提出一种新的二进制癌症驱动通路识别方法PEA-BLMWS。首先,利用已有的基因表达数据,通过对比正常基因与突变基因表达量的差异,挖掘潜在的基因突变数据;其次,引入蛋白质相互作用网络数据,构建出一个改进的二进制线性最大权重子矩阵模型;最后,提出一种双亲协同进化算法求解该矩阵模型。在GBM(glioblastoma)和OVCA(ovarian cancer)数据集上的实验结果表明,相比于其他先进的Dendrix、CCA-NMWS和CGP-NCM识别方法,PEA-BLMWS识别的基因集中有更多基因富集在已知的信号通路中,未富集在信号通路中的基因也与癌症的发生密切相关,故该识别方法可作为一种驱动通路识别的有效工具。
关键词:驱动通路; 基因突变; 基因表达; 进化算法
中图分类号:TP391 文献标志码:A
文章编号:1001-3695(2024)06-018-1728-07
doi:10.19734/j.issn.1001-3695.2023.10.0497
Binary cancer single-driver pathway identification model and algorithm
Abstract:The researches on driver pathway identification in cancer rely on traditional biological experiments, which have the drawbacks of being time-consuming, labor-intensive and costly. This paper proposed a novel binary cancer driver pathway identification method called PEA-BLMWS(parental evolutionary algorithm-binary linear maximum weight sub-matrix). Firstly, it utilized the existing gene expression data to uncovered potential gene mutation data by comparing the differences in expression levels between normal and mutated genes. Secondly,it incorporated protein-protein interaction network data to construct an improved binary linear maximum weight sub-matrix model. Finally,it proposed a parental evolutionary algorithm to solve this matrix model. Experimental results on the GBM(glioblastoma) and OVCA(ovarian cancer) datasets show that compared to other advanced identification methods such as Dendrix, CCA-NMWS and CGP-NCM, the gene set identified by PEA-BLMWS has more genes enriched in known signaling pathways, and genes not enriched in signaling pathways are also closely related to the occurrence of cancer. Therefore, this identification method can serve as an effective tool for driving pathway identification.
Key words:driver pathway; gene mutation; gene expression; evolutionary algorithm
0 引言
癌症的产生和发展与基因组的突变密切相关,且癌症高度受体细胞基因突变驱动,体细胞基因突变的类型主要包括单核苷酸变异、拷贝数变异以及核苷酸序列重复、插入以及缺失等[1]。并不是所有的基因突变都能导致癌症的发生,只有少部分突变能够使细胞具有生长优势,进而对癌症的产生与发展起到促进作用,这种突变称之为驱动突变[2]。对癌症发展起促进作用的基因称为驱动基因[3]。癌症细胞中驱动基因的集合称作驱动通路。不同的驱动基因可能共同作用于相同的生物学通路,生物学通路是指在生物体细胞内,一系列生物化学分子通过多个级联反应以实现一系列特定功能的路径。驱动通路中的一个基因发生突变时,可能干扰整个生物学通路的调控机制,从而促使癌症的发生[4]。故将关注点转移到生物学通路水平上,可以帮助科学家更加了解驱动通路与癌症的靶向关系,对于研究癌症的发病机理以及开发抗癌药物具有重要意义[5]。然而,通过生物实验进行驱动通路的识别需要对大量基因进行测序,花费大量时间并且经济成本高昂。随着高通量测序技术的发展,许多大型规模癌症基因测序项目得以开展,例如癌症基因组图谱计划(The Cancer Genome Atlas,TCGA)[6]和国际肿瘤基因协作组(International Cancer Genome Consortium,ICGC)[7]。通过进行基因测序项目,本文获取了大量的多组学数据,这为利用计算方法来识别驱动通路提供了可能。与传统的生物实验相比,使用计算方法来识别驱动通路具有高效率和低成本的优势[8]。
1 相关研究
根据识别的驱动通路数量的不同,现有的驱动通路识别问题分为单驱动通路识别[9]和协同驱动通路识别[10]两大类。协同驱动通路识别融入了基因、miRNA等相关组学数据,能够更加全面地分析癌症发生的机制,但存在处理数据量大,模型计算难度大等问题[11,12]。与协同驱动通路识别相比,尽管单一驱动通路识别方法仅考虑与基因相关的信息,会忽略一些在癌症发生中具有重要作用的组学数据,如miRNA等,导致突变矩阵融入的数据类型受限,但这种方法可快速确定致癌基因,从而为癌症治疗提供可靠依据。两类识别方法都存在各自的优缺点,本文聚焦单驱动通路识别问题进行研究。
2012年,Vandin等人[9]首次提出驱动通路具备高覆盖度和高互斥度两个重要性质。高覆盖度是指大量患者样本被驱动基因覆盖;高互斥度是指在一个癌症患者样本中,同一驱动通路中的基因同时发生突变的概率很小。其提出的最大权重子矩阵(maximum weight submatrix,MWS)问题模型是后续驱动通路识别方法的基础。对应求解MWS问题模型的基于马尔可夫链的求解方法Dendrix,仅使用突变数据识别癌症驱动通路,虽然这种方法在大多数情况下表现出良好的效果,但在搜索过程中可能会受限于局部最优解,无法跳出这种局限性。2012年,Zhao 等人[13]开发了一个软件包MDPFinder求解MWS问题模型,MDPFinder包括二进制线性规划方法(binary linear programming,BLP)和基于遗传算法的求解方法(genetic algorithm,GA)两种识别方法。BLP和GA针对不同类型的权重函数及其他组学数据均适用。MDPFinder的局限性是没有考虑覆盖度和互斥度的平衡,如果识别的两个基因集权重相同,其中覆盖度和互斥度更加平衡的基因集会被忽略。2016年,Zheng等人[14]整合基因表达数据和突变数据,提出一种基于遗传算法的多目标优化方法MOGA求解MWS问题模型。MOGA将基因表达数据融入MWS问题模型,通过调整模型中覆盖度和互斥度的权重,改进了MWS问题模型。相比之前方法,MOGA在调整高覆盖度和高互斥度之间的权衡方面更加可靠,但其局限性是未考虑到基因内部之间的联系。2021年,Wu等人[15]提出了一种新的识别模型和算法CGA-MWS,通过整合拷贝数变异、体细胞突变以及基因表达数据,构建了一个加权的非二进制突变矩阵,并重新定义了覆盖度和互斥度的概念。该模型的局限性在于它仅考虑了与基因相关的数据,而没有考虑其他可能的影响因素。2022年,Wu等人[16]提出了一种非线性最大权重子矩阵识别模型NMWS,基于覆盖度、互斥度以及网络连通性实现。NMWS 将癌症样本中的突变基因视为无向图中彼此连接的顶点,并通过图中顶点的聚集度来衡量驱动通路的互斥性,然后提出一种竞争协同演化算法CCA来求解NMWS模型。但NMWS模型只研究了基因集内部相互之间的联系,对其他网络信息考虑不全面。2023年,Zhu等人[17]提出了一种新的癌症驱动通路识别方法CGP-NCM,该方法在不预先设置人工参数的情况下构建了一个加权的非二进制突变矩阵,并提出了一种无参数的蛋白质相互作用网络识别模型,最后,设计了一种基于粒子群优化算法的协同进化算法,用于求解该模型。
此外,单一组学数据存在噪声且包含的信息过于独立,导致基于单一组学数据的驱动通路识别得到的普遍不是最优解。因此,针对现有单驱动通路识别方法的局限性,本文提出了一种新的二进制癌症驱动通路识别方法PEA-BLMWS。首先,整合体细胞突变数据、拷贝数变异数据和基因表达数据,生成一个二进制突变矩阵;其次,引入蛋白质相互作用网络数据,构建一个改进的二进制线性最大权重子矩阵模型(binary linear maximum weight submatrix,BLMWS),BLMWS模型充分考虑了驱动通路的高覆盖度和高互斥度的特性、突变基因对其他未突变基因的影响、蛋白质网络连通性等因素,提高了基因在样本中的突变可信度;最后,提出一种改进的双亲进化算法PEA(parental evolutionary algorithm)求解BLMWS模型。实验结果表明,本文提出的PEA-BLMWS可快速高效地识别出多种具有生物学意义的单驱动通路。
2 BLMWS模型构建
2.1 矩阵定义
使用计算的方法识别驱动通路,需要定义一组形式化表述的矩阵符号,对应表示多种组学数据。一组癌症样本集合表示为P,数量为|P|。一组备选基因集合表示为G,数量为|G|。
体细胞突变矩阵S|P|×|G|,矩阵元素的值为sij∈{0,1}(i=1,2,…,|P|,j=1,2,…,|G|),如果第j个基因在第i个癌症样本中发生突变,则sij=1,若无突变,则sij=0。
拷贝数变异矩阵C|P|×|G|,矩阵元素的取值范围为cij∈{-2,-1,0,1,2}(i=1,2,…,|P|,j=1,2,…,|G|),cij表示第j个基因在第i个癌症样本中的拷贝数变异值。
基因表达矩阵E|P|×|G|,矩阵元素值为实数,矩阵元素eij表示第j个基因在第i个癌症样本中的表达量,即eij∈R(i=1,2,…,|P|,j=1,2,…,|G|)。
蛋白质互作矩阵Q|G|×|G|,矩阵的行和列均代表基因,其中矩阵元素qij∈{0,1}(i=1,2,…,|G|,j=1,2,…,|G|,i≠j),qij=1表示基因i与j有相互作用,qij=0则表示两个基因无相互作用。
突变矩阵D|P|×|G|,体细胞突变矩阵S|P|×|G|与拷贝数变异矩阵C|P|×|G|的交集称为突变矩阵,即矩阵D|P|×|G|=S|P|×|G|∩C|P|×|G|。其中矩阵元素dij∈{0,1}(i=1,2,…,|P|,j=1,2,…,|G|),若sij和cij取值都为1,则dij=1,反之,则dij=0。
倍数矩阵T|P|×|G|,矩阵元素tij(i=1,2,…,|P|,j=1,2,…,|G|)的值与基因表达矩阵E|P|×|G|的矩阵元素eij有关。设基因表达矩阵E|P|×|G|中的第j个基因的所有正常样本的基因表达量的平均值为,则矩阵元素tij=eij/。
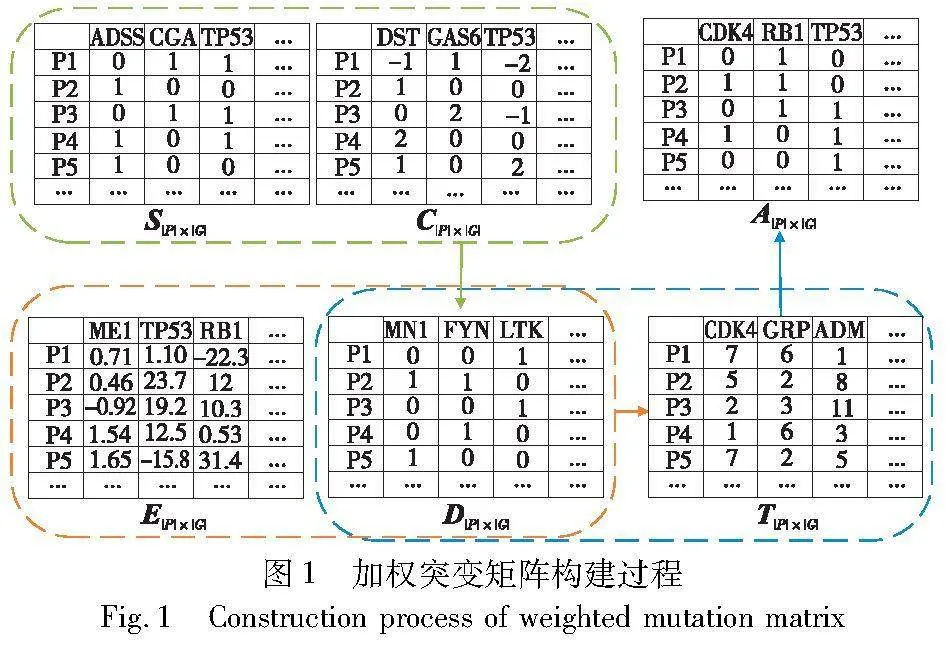
加权突变矩阵A|P|×|G|,为了提高未突变基因的潜在突变可能性,对矩阵D|P|×|G|中的未突变基因进行突变,提升了潜在突变基因的受关注度,得到一个加权突变矩阵A|P|×|G|。矩阵元素aij的值如式(1)所示。
其中:λ是用于确定是否进行突变操作的倍数阈值,经大量实验验证,λ的最优值为3。加权突变矩阵A|P|×|G|的具体构建过程,如图1所示。
2.2 通路特性定义
根据先前的研究,癌症驱动通路最大子矩阵模型具备高覆盖度和高互斥度两个重要特征。本文在基因相关数据和蛋白质相互作用的数据基础上,引入相关度和连通性的概念或指标,将癌症驱动通路的识别模型转换为一个多目标优化问题。在这个问题中,通过参数K2来减弱基因相关度和蛋白质连通性对驱动通路的影响,以优化模型的性能。
2.2.1 覆盖度
设矩阵A|P|×|G|的任意一个子矩阵为M|P|×K(1<K<|G|),当aij=1(i=1,2,…,|P|)时,令Γ(j)表示矩阵M|P|×K中第j个基因发生突变的样本集合。令CO(M)表示矩阵M|P|×K的覆盖度,定义如式(2)所示。
2.2.2 互斥度
假设p和q代表矩阵M|P|×K中任意两个基因(p≠q),当aip=1且aiq=1时,令Γ(pq)表示p和q同时发生突变的样本集合。令MU(M)表示矩阵M|P|×K的互斥度,定义如下:
2.2.3 相关度
2.2.4 连通性
令PE(M)表示蛋白质互作矩阵Q|G|×|G|中基因之间的连通性,定义如式(5)所示。
2.3 构建过程
基于上述定义,定义一个函数W(M),在给定的加权突变矩阵A|P|×|G|中寻找使W(M)函数值最大的子矩阵M|P|×K,W(M)的定义如式(6)所示。
W(M)=CO(M)+MU(M)+RE(M)+PE(M)(6)
因此,对应使函数W(M)取得最大值的矩阵M|P|×K中所包含的基因组就是驱动通路识别问题的解。识别癌症驱动通路问题转换为求解使函数W(M)取得最大值的子矩阵M|P|×K。
3 PEA算法构建
3.1 染色体编码方案
在遗传算法中,种群是指用遗传算法解决问题时,初始时给定的多个解的集合。初始时设置两个种群,每个种群包含n条染色体,每条染色体代表问题的一个解。染色体编码一般有十进制编码和二进制编码两种方式。本文种群中的染色体采用十进制编码方式,在备选基因集合G中随机挑选K个基因,生成初始染色体X={x1,x2,…,xK}(xi∈G,i=1,2,3,…,K)。
3.2 适应度函数
适应度函数是一种用来对种群中任意个体(染色体)的环境适应性进行度量的函数,其函数值是遗传算法实现优胜劣汰的依据。设每条染色体X代表子矩阵M|P|×K中的K个基因,对应的适应度函数F(X)定义如下:
F(X)=W(M)(7)
3.3 选择算子
选择算子的作用是对种群中的个体进行优胜劣汰操作,根据个体适应度函数值从种群中选出优秀的个体,传给下一代种群。为了避免算法陷入过早收敛,选择算子使用轮盘赌选择法、锦标赛选择法以及精英策略三种方法结合的方式实现以下的流程:
a)对于每一个种群,精英策略直接将种群中最优个体从父代传递到子代;
b)用轮盘赌和锦标赛选择法从两个种群中各选择一半个体进入子代。
以上三种方法相结合实现的选择算子能够明显增加搜索的广度,虽然算法的收敛速度略有降低,但总体效果较好。
3.4 交叉算子
交叉算子的目的是增强算法的局部搜索能力,对应的实现流程如下:
3.5 变异算子
变异算子可以增加种群的多样性,避免算法过早陷入局部最优解,本文设计了单点变异算子和概率变异自适应算子来增加种群的多样性。
3.5.1 单点变异算子
对于染色体中的基因,执行基于贪心策略的单点变异算子,具体流程如下:
a)给定一个染色体X,随机删除X中一个基因得到染色体X′,对应的候选基因集为G′={g|g∈G,gX};
b)从候选基因集G′中随机挑选一个基因加入到X′中;
c)比较F(X)与F(X′),如果执行单点变异后的染色体的适应度小于变异前染色体适应度,则重复执行步骤b)。若候选基因集G′中的基因都不满足F(X)<F(X′),则染色体X保持不变,否则得到变异后的新染色体X′。
3.5.2 概率变异自适应算子
对于种群中的染色体,采用概率变异算子实现自适应的改变,具体流程如下:
a)种群平均适应度是指该种群内所有个体适应度值的平均值,设任一种群的种群平均适应度为average_fit,设有该种群任一染色体X,则其适应度值为F(X)。
b)若F(X)≥average_fit,则该染色体突变基因数量为大于等于0.1×K的最小整数;若F(X)<average_fit,则该染色体突变基因数量为小于等于0.5×K的最小整数;设最终步骤b)染色体突变数量为temp。
d)从候选基因集G′中随机挑选temp个基因加入到X′中,若X=X′,则重复执行步骤d),直到得到多点变异后的新染色体X′。
3.6 算法流程
算法:PEA
4 实验与分析
4.1 实验设置
4.1.1 数据集预处理
对胶质母细胞瘤(GBM)和卵巢癌(OVCA)两种原始的真实癌症数据集进行相应的预处理,蛋白质互作网络数据取自文献[19]。
a)原始胶质母细胞瘤数据集包括91个患者样本的体细胞突变数据,206个患者样本的拷贝数变异数据以及259个患者样本的基因表达数据。对冗余数据处理后,得到覆盖90个患者样本的440个基因的基因突变矩阵和基因表达矩阵,组成实验所用的新胶质母细胞瘤(GBM)数据集。
b)原始卵巢癌数据集包括313个患者样本的突变数据,以及489个患者样本的基因表达数据。对冗余数据处理后,得到覆盖313个患者样本的2 547个基因的突变矩阵和基因表达矩阵,组成实验所用的新卵巢癌(OVCA)数据集。
4.1.2 参数设置
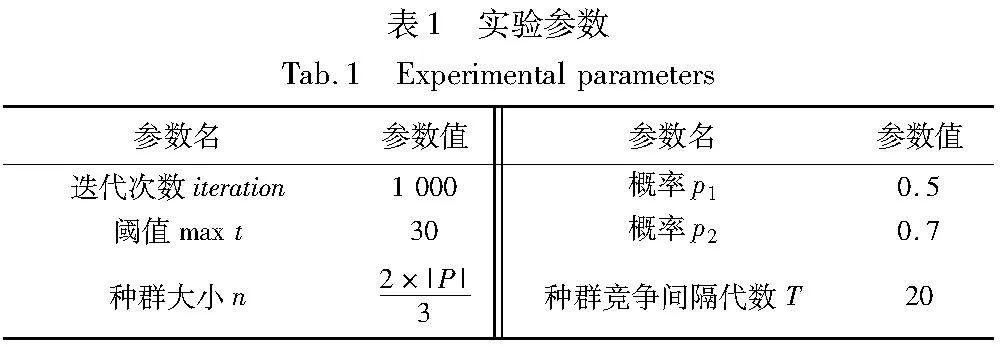
经大量实验验证后,识别方法PEA-BLMWS对应的超参数值设置如表1所示。
4.1.3 实验环境
实验在一台电脑(AMD Ryzen 7 5800H,3.20 GHz,内存16 GB)上进行,编译运行工具为R 4.2.1。
4.2 实验结果
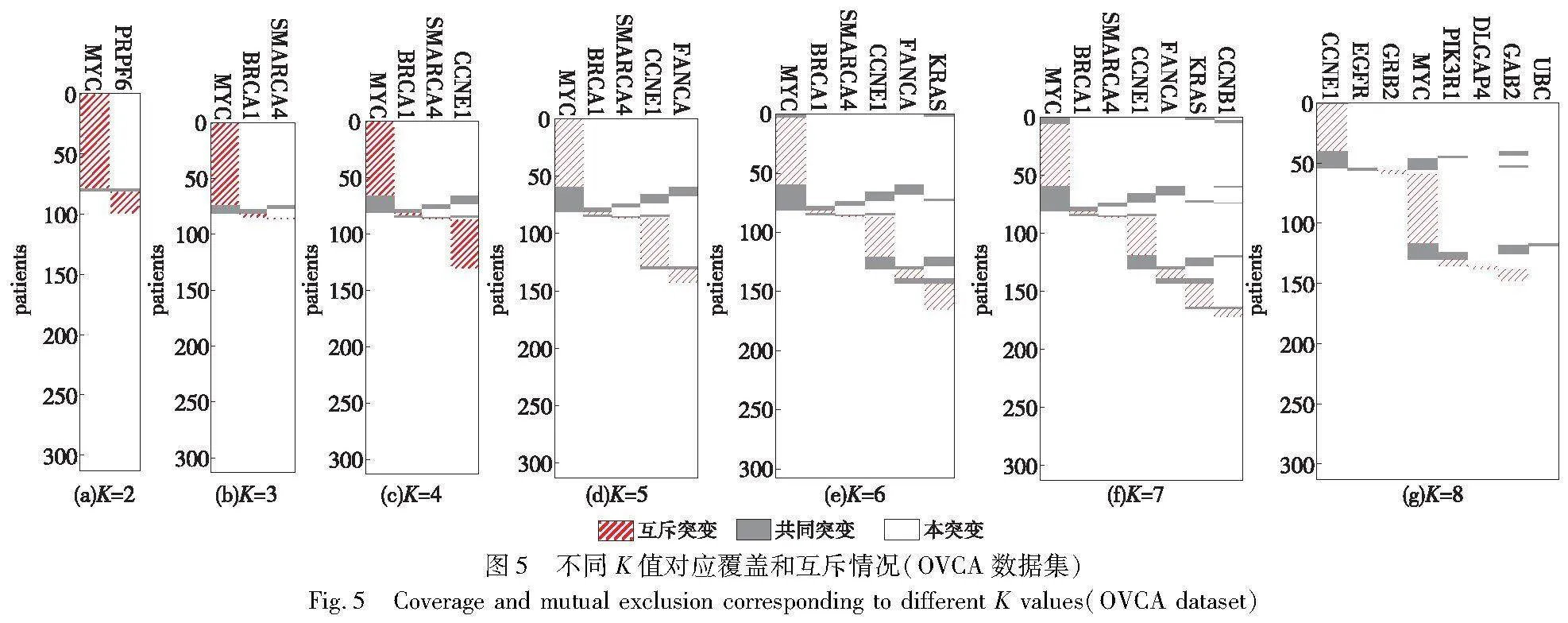
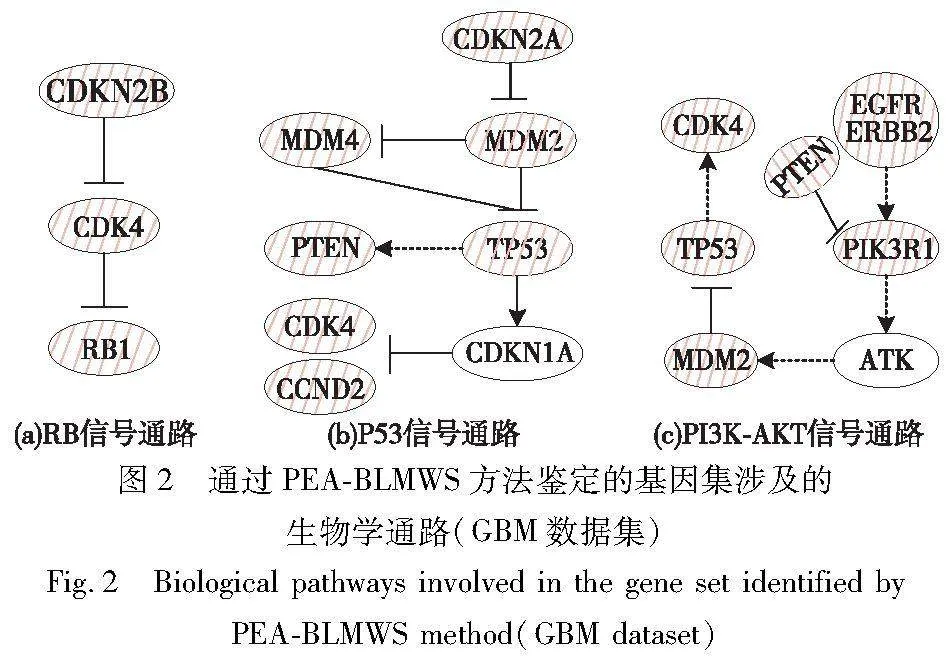
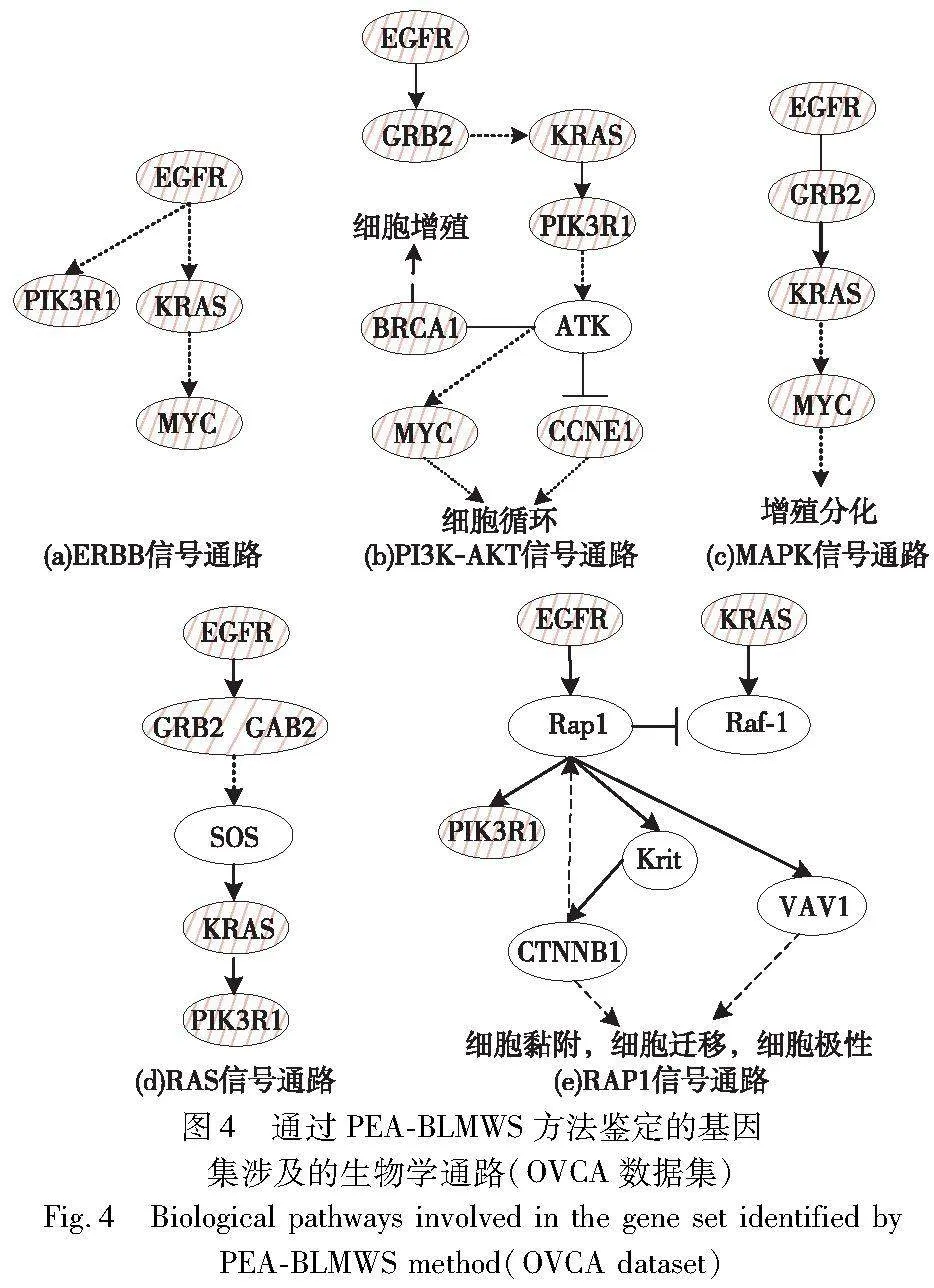
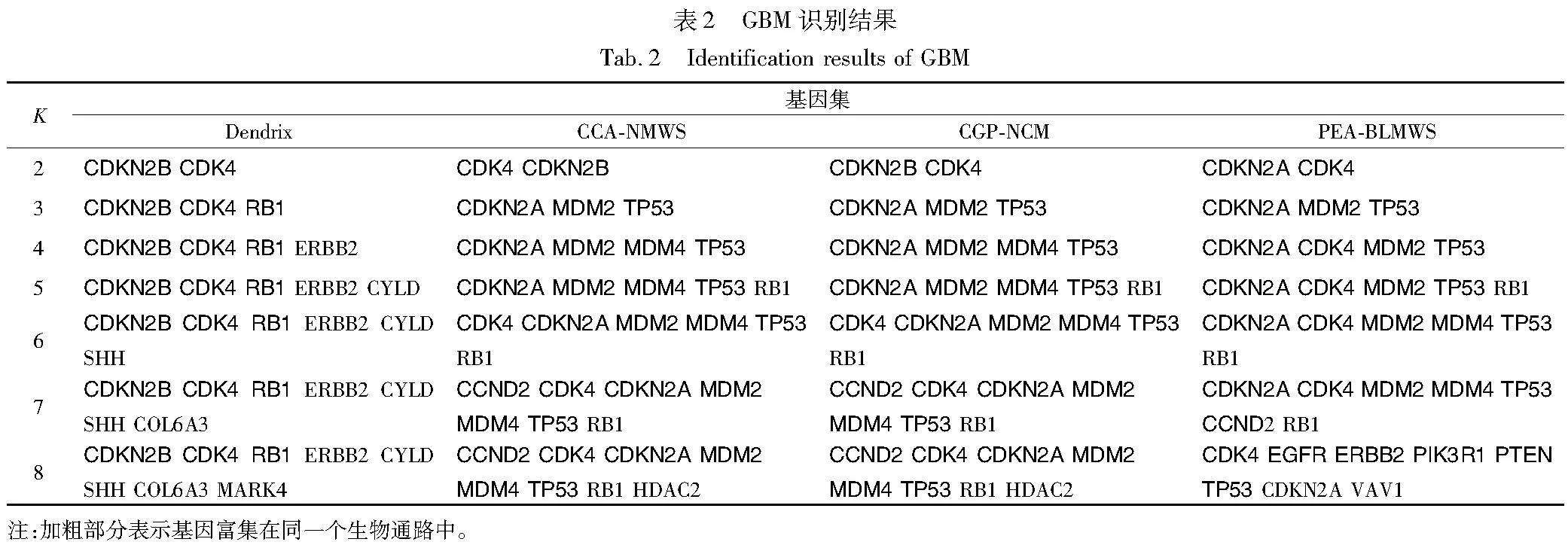
设置参数K从2变化到8,将三种基准方法(Dendrix、CCA-NMWS、CGP-NCM)和PEA-BLMWS方法识别到的基因集作比较,GBM数据集识别的结果见表2,OVCA数据集识别的结果见表3。图2和4为基因集对应信号通路图,图中实线表示抑制作用,实线箭头表示直接激活作用,虚线箭头表示间接激活作用,有图案填充的椭圆代表识别的基因,无填充的椭圆表示与识别到的基因相关联的基因。
4.2.1 GBM数据集实验结果
GBM识别结果如表2所示。结果分析如下:
a)基因集分析
①当K=2时,如图2(a)所示,三种基准方法识别到的基因集(CDKN2B CDK4)都富集在RB信号通路中, PEA-BLMWS可以识别出其他方法未识别到的基因集(CDKN2A CDK4), (CDKN2A CDK4) 是P53信号通路的一部分。
②当K=3时,如图2(a)所示,Dendrix识别的基因集(CDKN2B CDK4 RB1)富集在RB信号通路中。如图2(b)所示,CCA-NMWS、CGP-NCM、PEA-BLMWS识别的基因集(CDKN2A MDM2 TP53) 同样富集在P53信号通路中。相关研究表明,MDM2基因在P53信号通路中失调会抑制P53信号通路在抑制癌症方面的作用[20]。
③当K=4时,如图2(b)所示,CCA-NMWS和CGP-NCM识别到的基因集(CDKN2A MDM2 MDM4 TP53)是P53信号通路的一部分。PEA-BLMWS识别到不同的基因集(CDKN2A CDK4 MDM2 TP53),该基因集同样富集在P53信号通路中。P53信号通路的失调与胶质母细胞瘤细胞的增殖、侵袭、转移、凋亡以及癌细胞干性有关,P53蛋白的致癌变体对胶质母细胞瘤有促进作用[21]。
④当K=5时,如图2(b)所示,CCA-NMWS、CGP-NCM和PEA-BLMWS识别的基因集中,只有基因RB1与其他基因没有富集在同一个信号通路中。RB1是一个重要的癌基因,当其表达产物超过正常水平时会加快细胞增殖速度并降低细胞活性[22]。
⑤当K=6、7时,如图2(b)所示,除了基因RB1,CCA-NMWS、CGP-NCM与PEA-BLMWS识别到的基因集均富集在P53信号通路中。基因MDM4与其他三种基因CDKN2ATP53和MDM2共同参与P53信号通路。基因MDM4与MDM2具有相似的结构和功能,它们都在P53信号通路中充当致癌抑制剂以抑制肿瘤活性[21]。
⑥当K=8时,如图2(c)所示, PEA-BLMWS识别到一个不同的基因集(CDK4 EGFR ERBB2 PIK3R1 PTEN TP53),该基因集富集在PI3K-AKT信号通路中。PTEN基因在抑制肿瘤的产生和发展中起到重要作用,当PTEN基因发生缺失或突变时,会导致细胞增殖加速,并减缓细胞的凋亡速度。这些变化可以促进胶质母细胞瘤的发展,PTEN和TP53的失活会对胶质母细胞瘤的发展起到促进作用[23]。
b)通路特性分析
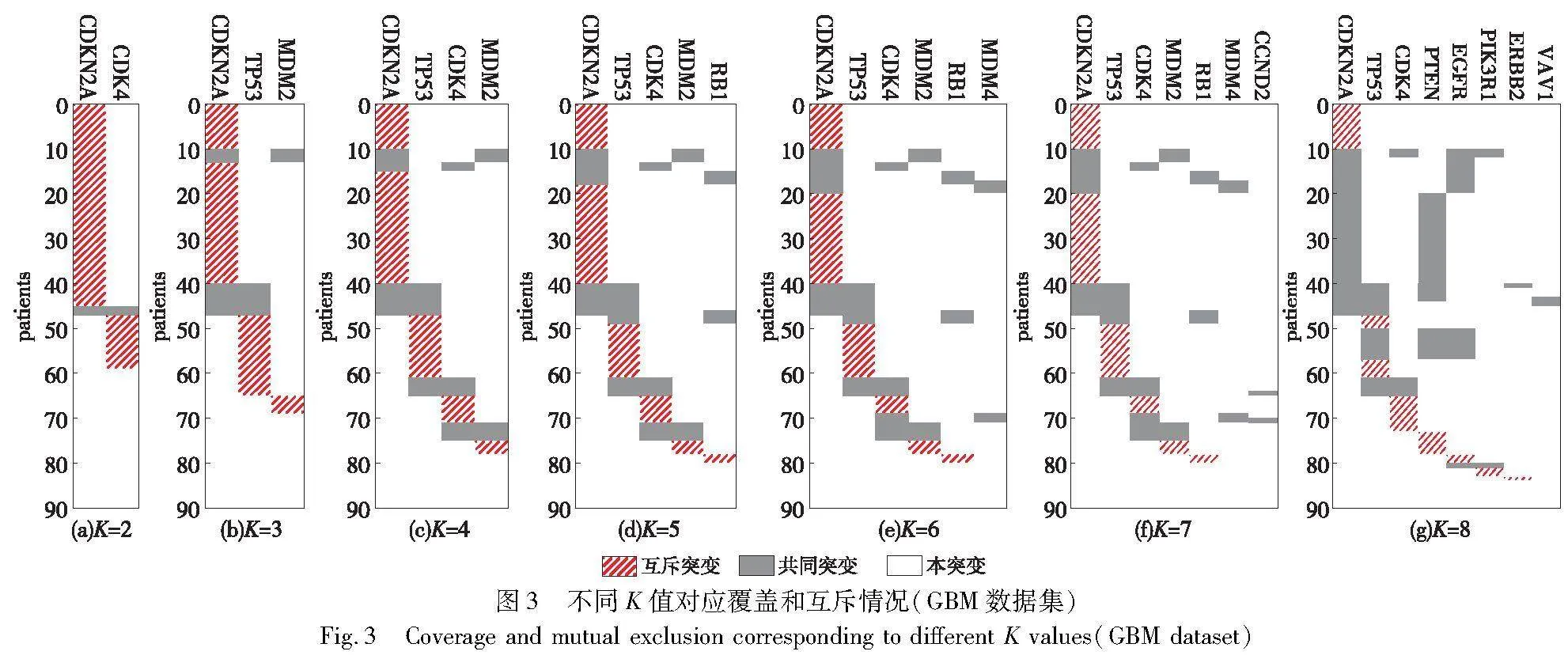
在胶质母细胞瘤癌症样本中,本文PEA-BLMWS方法识别的基因集在不同K值的覆盖和互斥下的情况如图3所示。PEA-BLMWS方法在基因集的覆盖度方面表现出色。它能够覆盖绝大多数样本中的突变基因,并且绝大部分突变基因之间呈现互斥的特点。驱动通路具有高覆盖度和高互斥度这两个重要性质,因此,根据覆盖度和互斥度的表现,可以认为PEA-BLMWS方法在GBM数据集上表现良好。
4.2.2 OVCA数据集实验结果
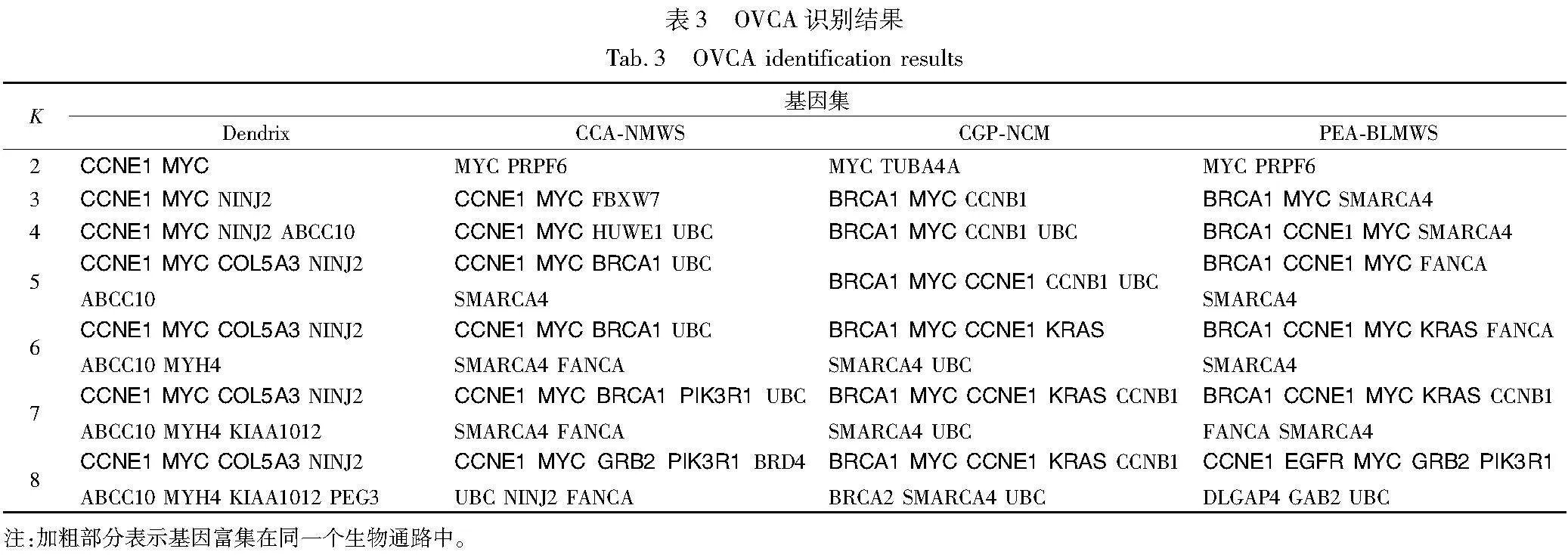
OVCA识别结果如表3所示。结果分析如下:
a)基因集分析
由表3可知,当K=2,3,4时,Dendrix识别的基因集只有2个基因富集在同一信号通路,当K=5,6,7,8时,Dendrix识别的基因集只有3个基因富集在同一信号通路。与CCA-NMWS、CGP-NCM和PEA-BLMWS方法相比, Dendrix方法识别的基因集中富集在同一信号通路中的基因较少。
①当K=3时,如图4(b)所示,PEA-BLMWS识别到的基因集中,有2个基因(BRCA1 MYC)富集在PI3K-AKT信号通路。PI3K-AKT信号通路在各种癌细胞的迁移中起着重要作用,研究表明,至少15%的侵袭性卵巢癌是由基因BRCA1和BRCA2突变引起的[24]。SMARCA4基因是一个重要的基因,SMARCA4表达缺失会造成卵巢高钙血症小细胞的癌变[25]。
②当K=4时,如图4(b)所示,在PEA-BLMWS识别的基因集中,有3个基因(BRCA1 CCNE1 MYC)富集在PI3K-AKT信号通路,而其他三种方法识别的基因集只有2个基因富集在PI3K-AKT信号通路。
③当K=5时,如图4(b)所示,CCA-NMWS、CGP-NCM与PEA-BLMWS识别的基因集中,有3个基因(BRCA1 CCNE1 MYC)富集在PI3K-AKT信号通路中。其中新识别的基因FANCA具有肿瘤抑制作用,其等位基因突变可能增加卵巢癌的患病几率[26]。当K>4时,Dendrix识别的基因集有且只有3个基因富集在同一信号通路,富集在同一信号通路中基因的数量并没有随着K的增大而增加。
④当K=6时,如图4(b)所示,CCA-NMWS方法识别的基因中只有3个基因富集在同一个信号通路。如图4(a)(c)所示,CGP-NCM与PEA-BLMWS识别的基因中有4个基因(BRCA1 CCNE1 MYC KRAS)富集在PI3K-AKT信号通路中,其中基因(MYC KRAS)也在MAPK以及ERBB信号通路中富集。MAPK信号通路在卵巢癌发生中起重要作用,并参与卵巢癌细胞的迁移[27]。具有YAP的ERBB信号通路形成自分泌环以诱导卵巢癌的发生和发展[28]。
⑤当K=7时,如图4(b)所示,CCA-NMWS、CGP-NCM与PEA-BLMWS方法识别的基因集有4个基因富集在同一个信号通路中。当K=8时, PEA-BLMWS识别的基因集在信号通路富集情况优于其他方法。CCA-NMWS与CGP-NCM方法识别到的基因集只有4个基因富集在同一信号通路,如图4(b)所示。PEA-BLMWS识别到的基因集有5个基因(CCNE1 EGFR GRB2 MYC PIK3R1)富集在PI3K-AKT信号通路。如图4(c)~(e)所示,基因(EGFR GRB2 MYC)富集在MAPK信号通路中;基因(EGFR GRB2 GAB2 PIK3R1)富集在RAS信号通路中;基因(EGFR PIK3R1)富集在RAP1信号通路中。RAS信号通路在驱动细胞增殖中起着重要作用,该信号通路的失调通常发生在肿瘤发生过程中[29]。RAP1通路通过促进MMP-2和MMP-9的分泌来调节卵巢癌的侵袭和转移[30]。
b)通路特性分析
在卵巢癌样本中,PEA-BLMWS方法识别的基因集在不同K值的覆盖和互斥下的情况如图5所示。它能够覆盖大多数样本中的突变基因,且绝大部分突变基因之间呈现互斥的特点。因此,可以认为PEA-BLMWS方法在OVCA数据集上表现良好。
4.3 显著性检验
为了评估实验结果的显著性并衡量其与理论最优值之间的偏离程度,本文采用了随机检验方法。设识别到的K个基因的函数值为W(M),W(Mi)为随机选取的K个基因的函数值,重复随机挑选1 000次,统计W(Mi)>W(M)的次数,显著性检验的公式如下:
经大量实验,p=0.04时,实验结果是显著的。
5 结束语
本文提出了一种单驱动通路识别方法,不依赖于先验知识,而是采用从头识别的方法。该方法基于体细胞突变数据、拷贝数变异数据和基因表达数据构建了一个加权的二进制突变矩阵。在确保驱动通路具有高覆盖度和高互斥度的基础上,引入了基因之间的相互作用和蛋白质互作网络,构建了一个新的识别模型。为了解决该模型,提出了一种双亲遗传算法PEA。该算法使用锦标赛选择法和轮盘赌选择的组合作为选择算子,并基于贪心策略设计了交叉算子,从而能够高效地找到最优解。最后,通过在胶质母细胞瘤和卵巢癌数据集上进行实验,实验结果表明,与其他基准方法相比,本文PEA-BLMWS在识别基因集时能够更好地捕捉到富集在已知信号通路中的基因,而那些在信号通路中没有富集的基因已经被证实与癌症的发生和发展密切相关。为了进一步提升模型的性能和可靠性,本文计划在更大规模的数据集上展开实验。同时,本文也将结合当前机器学习和深度学习的知识,探索可能存在于驱动通路中的其他特征,以进一步完善驱动通路识别模型。
参考文献:
[1]Greenman C, Stephens P, Smith R,et al. Patterns of somatic mutation in human cancer genomes[J]. Nature, 2007,446(7132):153-158.
[2]Hou J P, Ma Jian. DawnRank: discovering personalized driver genes in cancer[J]. Genome Medicine, 2014,6: article No.56.
[3]Ding Li, Getz G, Wheeler D A, et al. Somatic mutations affect key pathways in lung adenocarcinoma[J]. Nature, 2008,455(7216): 1069-1075.
[4]Hahn W C, Weinberg R A. Modelling the molecular circuitry of can-cer[J]. Nature Reviews Cancer, 2002,2(5): 331-341.
[5]Boca S M, Kinzler K W, Velculescu V E, et al. Patient-oriented gene set analysis for cancer mutation data[J]. Genome Biology, 2010,11(11): article No.R112.
[6]Hudson T J, Anderson W, Aretz A, et al. International network of cancer genome projects[J]. Nature, 2010,464(7291): 993-998.
[7]Chin L, Andersen J N, Futreal P A. Cancer genomics: from disco-very science to personalized medicine[J]. Nature Medicine, 2011,17(3): 297-303.
[8]Zhang Junhua, Zhang Shihua. The discovery of mutated driver pathways in cancer: models and algorithms[J]. IEEE/ACM Trans on Computational Biology and Bioinformatics, 2016,15(3): 988-998.
[9]Vandin F, Upfal E, Raphael B J. De novo discovery of mutated dri-ver pathways in cancer[J]. Genome research, 2012,22(2): 375-385.
[10]Zhang Junhua, Wu Lingyun, Zhang Xiangsun, et al. Discovery of co-occurring driver pathways in cancer[J]. BMC Bioinformatics, 2014,15(1): 271.
[11]Wang Jun, Yang Ziying, Domeniconi C, et al. Cooperative driver pathway discovery via fusion of multi-relational data of genes, mi-RNAs and pathways[J]. Briefings in Bioinformatics, 2021,22(2): 1984-1999.
[12]Yang Ziying, Yu Guoxian, Guo Maozu, et al. CDPath: cooperative driver pathways discovery using integer linear programming and Markov clustering[J]. IEEE/ACM Trans on Computational Bio-logy and Bioinformatics, 2019,18(4): 1384-1395.
[13]Zhao Junfei, Zhang Shihua, Wu Lingyun, et al. Efficient methods for identifying mutated driver pathways in cancer[J]. Bioinformatics, 2012,28(22): 2940-2947.
[14]Zheng Chunhou, Yang Wu, Chong Yanwen, et al. Identification of mutated driver pathways in cancer using a multi-objective optimization model[J]. Computers in Biology and Medicine, 2016,72: 22-29.
[15]Wu Jingli, Zhu Kai, Li Gaoshi, et al. A model and algorithm for identifying driver pathways based on weighted non-binary mutation matrix[J]. Applied Intelligence, 2022,52(1): 127-140.
[16]Wu Jingli, Chen Xiaorong, Li Gaoshi, et al. A nonlinear model and an algorithm for identifying cancer driver pathways[J]. Applied Soft Computing, 2022, 129: 109578.
[17]Zhu Kai, Wu Jingli, Li Gaoshi, et al. A model and cooperative co-evolution algorithm for identifying driver pathways based on the integrated data and PPI network[J]. Expert Systems with Applications, 2023, 212: 118753.
[18]Cohen J, Chen Jingdong, Huang Yiteng, et al. Pearson correlation coefficient[M]//Noise Reduction in Speech Processing. Berlin: Springer, 2009: 1-4.
[19]Leiserson M D M, Vandin F, Wu H T, et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes[J]. Nature Genetics, 2015, 47(2): 106-114.
[20]Iwakuma T, Lozano G. MDM2, an introduction[J]. Molecular Cancer Research, 2003,1(14): 993-1000.
[21]Zhang Ying, Dube C, Gibert Jr M, et al. The p53 pathway in glioblastoma[J]. Cancers, 2018,10(9): 297.
[22]Goldhoff P, Clarke J, Smirnov I, et al. Clinical stratification of glioblastoma based on alterations in retinoblastoma tumor suppressor protein(RB1) and association with the proneural subtype[J]. Journal of Neuropathology & Experimental Neurology, 2012,71(1): 83-89.
[23]McLendon R, Friedman A, Bigner D,et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways[J]. Nature, 2008, 455(7216): 1061-1068.
[24]Pal T, Permuth-Wey J, Betts J A, et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases[J]. Cancer: Interdisciplinary International Journal of the American Cancer Society, 2005,104(12): 2807-2816.
[25]Moes-Sosnowska J,Szafron L,Nowakowska D,et al.Germline SMARCA4 mutations in patients with ovarian small cell carcinoma of hypercalcemic type[J]. Orphanet Journal of Rare Diseases, 2015,10: article No.32.
[26]Thompson E, Dragovic R L, Stephenson S A, et al. A novel duplication polymorphism in the FANCA promoter and its association with breast and ovarian cancer[J]. BMC Cancer, 2005,5: article No. 43.
[27]Ptak A, Hoffmann M, Gruca I, et al. Bisphenol a induce ovarian cancer cell migration via the MAPK and PI3K/Akt signalling pathways[J]. Toxicology Letters, 2014,229(2): 357-365.
[28]He Chunbo, Lyu Xiangmin, Hua Guohua, et al. YAP forms autocrine loops with the ERBB pathway to regulate ovarian cancer initiation and progression[J]. Oncogene, 2015,34(50): 6040-6054.
[29]Punekar S R, Velcheti V, Neel B G, et al. The current state of the art and future trends in RAS-targeted cancer therapies[J]. Nature Reviews Clinical Oncology, 2022,19(10): 637-655.
[30]Che Yaling, Luo Shujuan, Li Gang, et al. The C3G/Rap1 pathway promotes secretion of MMP-2 and MMP-9 and is involved in serous ovarian cancer metastasis[J]. Cancer Letters, 2015, 359(2): 241-249.
