难浮煤-极性捕收剂相互作用研究新视角:分子对接与诱导契合效应
2024-05-23蘧鹏程夏阳超薛志刚邢耀文桂夏辉
蘧鹏程 ,夏阳超 ,薛志刚 ,邢耀文 ,桂夏辉 ,田 佳
(1.中国矿业大学 化工学院, 江苏 徐州 221116;2.中国矿业大学 国家煤加工与洁净化工程技术研究中心, 江苏 徐州 221116;3.中南大学 资源加工与生物工程学院, 湖南 长沙 410083)
0 引 言
我国“富煤、贫油、少气”的资源禀赋和现阶段经济社会的发展实际决定了短期内仍离不开煤炭[1]。在“碳达峰、碳中和”的能源大背景下,煤炭行业的发展急需突破性技术,以更好地配合新能源发展,实现碳中和[2]。煤炭的开发利用随着开采机械化程度的提高,导致粉煤量占原煤的比率不断增大。浮选分离方法因其成本低、处理量大,是处理细粒煤分离提质的有效方法[3]。
浮选药剂是浮选技术变革的关键突破点之一,也是浮选过程强化的根本所在[4]。借鉴生物医药的思想见解来分析煤大分子与浮选药剂相互作用由来已久。浮选药剂专家见百熙认为药物作用的对象是人,浮选药剂作用的对象是矿物,二者之间具有互通性[5]。1894 年,FISHER 等[6]针对药剂的专一性提出了“锁钥模型”,认为药剂与靶体在相互作用之前其结构就有了互补性。在研究矿物-药剂分子相互作用过程中,很多科研工作者充分应用了“锁钥模型”研究药剂与矿物间作用,然后进行设计浮选捕收剂分子。秦伟[7]应用锁钥模型,解释了外来捕收剂在几何构型上与矿物表面的金属离子相匹配,则药剂与矿物更容易结合在一起,进而设计了11 种理论捕收剂性能较高的2-疏基苯丙咪唑类捕收剂。李文静[8]采用锁钥模型,明晰了药剂分子与矿物分子相互作用机制,根据构建的红柱石晶体模型对异羟肟酸类浮选药剂进行分子设计。
相比于矿物而言,我国煤泥成因复杂,分布环境多样,煤表面性质种类丰富,单种或常规浮选捕收剂在不同种类煤泥浮选过程中适应性较差[9],体现了浮选药剂的“专一性”较差,需要更深刻的理论模拟药物与矿物间的相互作用。KOSHLAND[10]于1958 年提出了“诱导契合理论”,认为药剂与靶体在空间上距离彼此接近时,不仅药剂分子会发生构型变化,靶体也会进行互补契合变化,该理论可为煤分子与药剂的相互作用研究提供借鉴。桂夏辉等[11-12]认为难浮煤浮选捕收剂的开发模式应由传统的经验性试探向精准设计转变,针对煤表面物化性质和难浮机理认知开发适配于不同种类难浮煤的高效浮选捕收剂筛选技术是当前浮选药剂研究的主要方向。这对药剂与煤分子间相互作用的研究提出了更高的要求,明晰极性药剂与煤分子间的微观作用行为可以为新型高效浮选药剂的开发打下坚实基础。
将药物分子筛选中的分子对接技术及诱导契合理论引入到难浮煤浮选极性捕收剂分子间相互作用研究中,以快速寻找对难浮煤表面结构具有靶向作用的极性烃类油捕收剂。选用原型分子(Protomol)技术表示煤大分子的活性区域,在活性区域中对小分子进行构象搜素,搜索出与煤分子贴合度最好、相互作用最强的药剂构型,在分子对接结果基础上开展基于诱导契合效应的煤大分子动力学优化,探究浮选捕收剂的作用机制及不同药剂间作用差异性机理。
1 试验材料与方法
1.1 难浮煤分子构建方法
难浮煤试验样品选自内蒙古神东矿区的低阶煤,表1 为难浮煤样品的工业分析和元素分析。分析煤样性质发现挥发分和氧元素含量较高,反映了低阶煤含有较多含氧官能团的特征。

表1 煤样工业分析和元素分析Table 1 Proximate analysis and elemental analysis of coal sample
采用傅里叶红外变换光谱仪(FTIR)对难浮煤表面化学结构性质进行分析(图1),将煤样全谱分为700~900 cm-1波段的芳香烃结构特征吸收峰、1 000~1 800 cm-1波段的含氧、氮、硫等杂原子官能团特征吸收峰、2 800~3 000 cm-1波段脂肪族结构特征吸收峰、3 000~3 600 cm-1波段的羟基结构特征吸收峰[13]。采用Origin 软件根据不同区段特征峰进行拟合并归属,后期将红外谱图数据结合其他分析手段得到煤大分子结构参数[14]。
采用CasaXPS 软件对煤样的X 射线光电子能谱图进行分峰拟合(图2),并根据文献对各拟合峰进行归属[15]。可发现碳的主要存在形式为芳香结构及取代烷烃碳;而氧的主要存在形式为羰基氧和醚或羟基类型氧,这些富含氧的强亲水性基团的存在是导致低阶煤等难浮煤难以浮选分离的根本原因[16]。

图2 煤样的XPS 谱图Fig.2 XPS spectrum peak of coal sample
煤样的13C-NMR 谱图反应煤中碳的存在形式及相对含量,根据煤样的化学位移,将煤样的13CNMR 谱图分为4 个区段[17]:化学位移为0~60 ppm的酯碳峰;化学位移为60~90 ppm 的醚氧峰;化学位移在90~165 ppm 的芳碳峰;化学位移在200 ppm的羰基碳峰。由于测量过程中的边带效应,原本在200 ppm 处的羰基碳峰整体偏移至174 ppm 左右。具体分峰拟合结果如图3 所示,根据各拟合峰的相对面积及半峰宽计算出可表征煤样性质的12 个结构参数,最终计算结果见表2。

图3 煤样的NMR 谱图Fig.3 NMR spectrum peak of coal sample

表2 煤样主要结构参数Table 2 Main structural parameters of coal samples
煤样的高分辨率透射电子显微镜结果图由Digtal Micrograph 软件及ImageJ 软件联合使用进行处理[18]。图4a 为煤样透射电镜原图,图4b 为圈定分析的部分,图4c—图4e 是对选定区域进行傅里叶变换-环形滤波过滤-反傅里叶变换等,图4f 为转换后谱图添加比例尺,使统计手绘条纹时方便获取条纹实际长度,图4g 将图片设定合适的阈值,对图片进行二值化处理,方便进行条纹的手绘操作。

图4 煤样的HRTEM 谱图处理流程Fig.4 HRTEM image processing of coal sampl
1.2 难浮煤分子结构模型
根据煤样的工业分析、元素分析、FTIR 和XPS分析等分析结果,构建神东低阶煤大分子结构,构建的神东煤分子式为C196H208N3O36,与元素分析结果最大差距为4.92%,具体比对数据见表3,可以认为构建的煤分子能够较好地代替试验用煤。在Material studio 软件中通过结构优化-能量优化-分子动力学模拟过程获得结构及能量稳定的煤大分子构型,为分子对接准备合适的对接受体,优化使用Forcite 模块下的COMPASS 力场,退火算法温度在800~298 K内实现煤大分子模型在NVT 系综的结构弛豫,优化后的煤大分子结果如图5 所示。难浮煤分子的13CNMR 计算曲线与实际曲线对比图如图6 所示。首先通过ACD/CNMR-Predictor 计算煤分子化学位移,将计算得到的化学位移导入GNMR 中计算得到模型的谱图。可以观察到计算曲线与实验曲线大致上可以获得拟合,这说明构建的神东煤分子在C 的存在形式上较为接近煤分子实际情况。

图5 难浮煤分子的优化构型Fig.5 Optimal configuration of difficult to flotation coal molecule
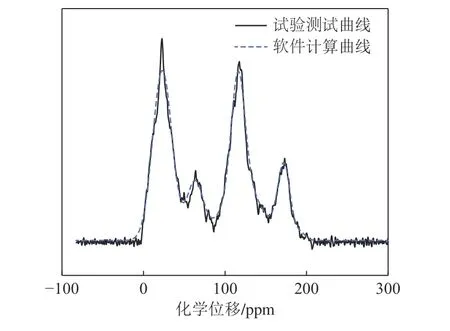
图6 难浮煤分子的13C-NMR 计算曲线与实际曲线对比Fig.6 Comparison of 13C-NMR calculation curve and actual curve of difficult floating coal molecules

表3 煤样和煤大分子结构的元素含量分析Table 3 Elemental analysis of samples and structural models
1.3 原型分子探针技术
采用原型分子探针技术确定煤分子活性位点并创建分子对接口袋的方法可以消除人为因素的影响,其原理为:采用N—H、C=O、单独氢原子为代表的空间型、氢键供体型和氢键受体型3 种探针,探测煤分子上与3 种探针相互作用较大的位点,这组探针在这些作用较强的位点形成 “煤分子补体”,能够填补煤分子内部和周围的大部分空隙。探针分子的选择与研究药剂种类无关,且不能够人为改变探针分子种类。
Protomol 原型分子算法主要原理为“锁钥模型”及“分子孔洞理论[19]”,主要分为“探针放置”“粘点识别”“口袋吸积”三步,即首先将3 种类型的探针分子密集的放置在煤分子周围,填充其表面,然后计算探针在表面的相互作用并进行过滤,只保留那些与煤分子相互作用强的探针。然后通过选择探针子集来定位粘点,这些探针集有可能在小体积内与煤分子产生强烈的累积相互作用,最后,一组球体在煤分子空腔内“膨胀”直到碰触到煤分子的范德华表面,将粘性点生长成对接口袋并输出排名靠前的对接口袋[19]。
1.4 捕收剂选择与优化
选取十二醇、十二醛、十二酸甲酯三种常规极性烃类油作为浮选捕收剂(表4),药剂的选择依据:油状液态、不易挥发、易获得、不含毒性或毒性较小。使 用Material studio 软 件 中 的Forcite 模 块 以及COMPASS 力场,对药剂分子进行结构-能量优化,为分子对接提供合适的捕收剂结构,图7 为优化后的捕收剂分子结构。

图7 优化后的捕收剂分子结构Fig.7 The optimized molecular structures of collectors

表4 试验捕收剂Table 4 Reagents used in experiments
1.5 分子对接方法
煤与捕收剂间的分子对接采用Sybyl 软件Docking Suite 模块完成,该模块是基于蒙特卡罗法在受体的活性区域产生构象,有效快速预测配体可能的结合模式,并对药剂与煤分子作用构型进行一致性打分,输出对接较好的分子构型[20]。和分子动力学和量子化学计算相比,分子对接考虑对接区域固定,极大地减少了采样空间和计算时间,适用于大批量药物甚至数据库的筛选,对煤炭浮选捕收剂的快速筛选意义重大。
分子对接筛选过程如图8 所示,首先采用Protomol 原型分子技术确定煤分子活性位点及药剂构象搜索区域,完成分子对接受体与配体对接约束条件之后,接着按照蒙特卡罗方法随机产生多个满足“空间匹配”、“能量匹配”要求的三维对接构象,依据一致性打分函数对三维对接构象进行打分并排序,输出对接结果较好的前几个捕收剂构型,完成煤与捕收剂间的分子对接。

图8 分子对接原理及诱导契合效应示意Fig.8 Schematic of molecular docking principle and induced fit effect
1.6 “诱导契合”模型
将分子对接后的药剂构型导出,采用Pymol软件将煤与药剂的分子对接结果整合为一个复合物,导入Materials Studio 软件。首先通过Modify 下面的Constraints 功能将药剂分子的三维空间进行固定,然后采用Forcite 模块,在COMPASS 力场下,对煤分子-药剂体系进行分子力学模拟,对煤分子-药剂复合物进行结构优化及能量优化,优化质量为精细,电荷采用立场分配,优化过程最大迭代步数为5 000 步。
1.7 浮选试验
选用XFDⅢ-1L 挂槽浮选机进行浮选试验,矿浆质量浓度为80 g/L,叶轮转速为1 800 r/min,刮板转速为33 r/min,充气量为0.1 m3/(m2·min),捕收剂用量为1 000 g/t,起泡剂用量为500 g/t。试验中首先将80 g 干燥煤样与自来水放置于浮选槽中,补齐液位混合调浆2 min,随后加入捕收剂搅拌2 min,加入仲辛醇继续搅拌30 s。试验备浆完毕后,充气10 s后开始收集精煤,时长共5 min。试验结束后,将精煤和尾煤样品过滤、烘干、烧灰并计算浮选精煤灰分和可燃体回收率。
2 结果与讨论
2.1 煤分子-药剂间的分子对接
在煤分子与药剂进行分子对接前,首先对煤分子活性区域进行检测,将氢原子、N—H 和C=三种探针原子与煤分子表面作用情况为依据,设定探针-煤分子相互作用强弱的阈值,对小于阈值的分子探针进行舍弃,将剩余合理的探针部分进行连接,最终成为煤分子的活性口袋,如图9a 灰色区域即为神东煤分子活性区域。以煤分子活性区域为依据,进行药剂-煤分子对接,对接结果如图9b—图9d 所示。通过分析对接结果发现十二酸甲酯呈直连结构贴合在煤分子活性区域中,而十二醇与十二醛分子距离活性区域中心较远,呈弯曲结构粘附在煤分子表面的活性位点附近。药剂的三维构型通过软件的“一致性打分”函数进行评价,十二酸甲酯与煤分子相互作用结合能最大为-23.655 5 kcal/mol,十二醛次之为-20.445 6 kJ/mol,十 二 醇 最 小 为-19.750 6 kJ/mol。对与煤分子“活性区域”贴合效果最好、作用效果最强的捕收剂分子给出较高的分值,一致性打分函数标准见文献[20]。

图9 煤分子活性区域及各种药剂最佳构型Fig.9 Diagram of coal molecular active region and optimal configuration of agents
2.2 药剂-煤分子间的诱导契合效应
煤大分子-药剂间的 “诱导契合”结果如图10所示,连接煤分子两端的C 原子,并以中间羟基为连接点,对煤分子的“契合程度”进行测量。十二醇煤分子的剩余角度为88.78°,十二醛煤分子的剩余角度为82.58°,这说明十二醇分子周围的煤分子“闭合程度”要小于十二醛周围煤分子的“闭合程度”,3 种药剂分子与煤分子的相互作用过程中,十二酸甲酯中煤分子的“闭合程度”最大,剩余的角度最小为81.93°。说明药剂分子与煤分子作用效果最强,这与分子对接得到的结合能呈现相同规律。
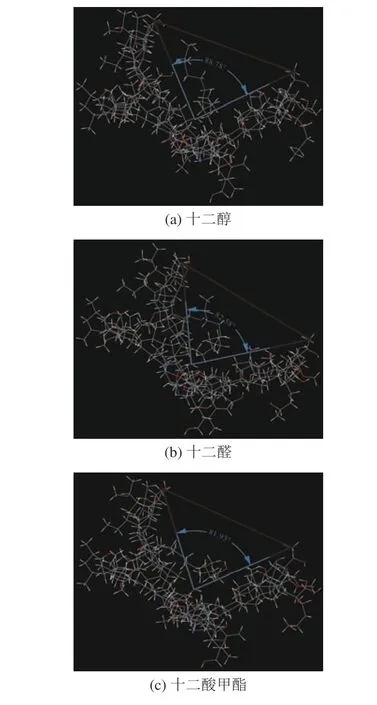
图10 煤分子与药剂分子的“诱导契合”图Fig.10 Coal molecule and pharmaceutical molecule induced coincidence diagram
2.3 极性捕收剂的浮选试验结果
采用分子对接中的3 种极性捕收剂对难浮煤进行浮选,结果如图11 所示。发现精煤可燃体回收率呈现出:十二醇<十二醛<十二酸甲酯的规律,其中十二酸甲酯和十二醛的浮选效果要远好于十二醇的浮选效果,十二酸甲酯可燃回收率高达85%左右,而十二醇的浮选效果较差,可燃体回收率在60% 左右。联系分子对接及诱导契合效应结果,结合能绝对值高、煤分子对药剂包裹程度大的捕收剂对应的浮选可燃体回收率更高,浮选试验验证了分子对接及诱导契合结果。

图11 十二醇、十二醛和十二酸甲酯捕收剂对难浮煤的浮选结果Fig.11 Flotation results of difficult-to-float coal with lauryl alcohol, lauraldehyde, and methyl laurate collectors
3 结 论
1)采用FTIR、XPS、HRTEM 等仪器分析手段分析煤样性质并构建煤大分子。基于Protomol 原型分子技术,能够较好的预测煤分子活性口袋,提供了一种煤分子活性口袋的确定方式。
2)采用煤分子为刚性、药剂分子为柔性的半柔性对接来探索药剂-煤分子间相互作用强弱,分子对接结合能强弱呈现十二酸甲酯>十二醛>十二醇的规律。根据分子对接构型分析,药剂能够较好地吸附在煤分子活性位点周围,进而达到提高煤分子疏水性的目的。以确定药剂分子最佳构型为依据,对煤-药剂间进行诱导契合效应的探索。探索发现十二酸甲酯周围的煤分子能够更好的契合十二酸甲酯的空间结构,煤分子表面与药剂表面更加贴合,酯类捕收剂导致煤分子收缩的角度变化最大,最终煤分子剩余角度为81.93°。
3)实际浮选结果呈现十二酸甲酯>十二醛>十二醇的规律,十二酸甲酯可燃体回收率约为85%,十二醛可燃体回收率约为80%,十二醇可燃体回收率最低,为60%。浮选结果与分子对接结合能及煤-药剂间诱导度呈现明显的正相关关系。论文为浮选捕收剂的快速筛选提供了技术途径,对未来环保型靶向浮选药剂的开发设计具有重要意义。
