光诱导基于EDA机理构建S-芳基黄原酸酯新策略
2024-05-15杨道山左峻泽
杨道山 左峻泽


主持人按语
专栏:光促进有机转化和应用
【特约主持人】 :张志国:“河南省杰出青年科学基金”入选者
100多年前,意大利化学家G. Ciamician 和 P. Silber意识到有机光化学反应具有重要意义.十几年前,当科学家将光氧化还原催化剂第一次应用到化学合成当中时,走向绿色化学的重大一步就这样迈出了.普林斯顿大学化学系的David MacMillan教授认为:“光氧化还原反应不仅仅让合成变得更快,更使以前根本不敢想的分子合成反应变为可能,而且你仅仅只需要一步反应”.光氧化还原催化剂主要包括金属和有机光催化剂.由于金属光催化剂当中的稀有金属造价不菲,近年来有机光氧化还原催化剂快速兴起.在对其修饰和设计之后可以达到廉价、高效催化氧化還原反应的效果,代替金属催化剂.如今,有机光化学与多学科、多领域高度交叉融合,其核心使命是发展高效、高选择性、高经济性、节能又环保的方式实现物质的化学转化.
本专栏围绕两种有机光催化剂促进的功能有机分子创制过程进行讨论,其中《光诱导基于EDA机理构建S-芳基黄原酸酯新策略》发展了通过光诱导的硫负离子与噻蒽盐间的电子给体受体过程实现C-S键构建的新策略.在温和的、无金属的条件下获得了一系列S-芳基黄原酸酯类化合物并实现了克级规模的制备,证明了反应的潜在应用价值,为黄原酸酯类化合物的合成提供了一种新思路.《一锅多组分反应合成3-(2-(芳基乙炔)苯基)喹唑啉酮》介绍了一种可用于通过自由基串联环化反应合成含氮杂环化合物的新型自由基受体化合物的构建方法.合成了一系列3-(2-(芳基乙炔)苯基)喹唑啉酮类化合物,该系列化合物可以作为自由基受体在蓝光诱导下转化为新型膦酰化喹唑啉酮并喹啉类化合物.
期待本专栏能够为从事有机光化学转化领域中有机功能分子的设计、合成和应用等相关领域的研究者提供参考.
摘 要:黄原酸酯类化合物作为药物化学、农业化学、材料化学中重要的组成部分,一直备受化学家们的关注.寻找简单、高效合成S-芳基取代黄原酸酯的方法引起了研究者广泛的关注.报道了一种可见光诱导下基于硫负离子与噻蒽盐间的EDA(电子供体受体)过程实现C-S键构建的新策略.在温和无金属的条件下,以中等至优良的收率得到了一系列S-芳基黄原酸酯,克级实验与日光实验证明了反应的应用潜力,为黄原酸酯类化合物的合成提供了一种新思路.
收稿日期:2023-06-27;修回日期:2023-08-13.
基金项目:国家自然科学基金(22271170).
作者简介(通信作者):杨道山(1982-),男,山东临沂人,青岛科技大学教授,主要从事有机合成方法学研究,E-mail:yangdaoshan@tsinghua.org.cn.
引用本文:杨道山,左峻泽.光诱导基于EDA机理构建S-芳基黄原酸酯新策略[J].河南师范大学学报(自然科学版),2024,52(3):1-9.(Yang Daoshan,Zuo Junze.A new strategy for the synthesis of S-arylxanthate via electron donor acceptor complex photoactivation[J].Journal of Henan Normal University(Natural Science Edition),2024,52(3):1-9.DOI:10.16366/j.cnki.1000-2367.2023.06.27.0001.)
关键词:黄原酸酯;EDA复合物;噻蒽盐;可见光诱导;C-S键的构建
中图分类号:O612 文献标志码:A文章编号:1000-2367(2024)03-0001-09
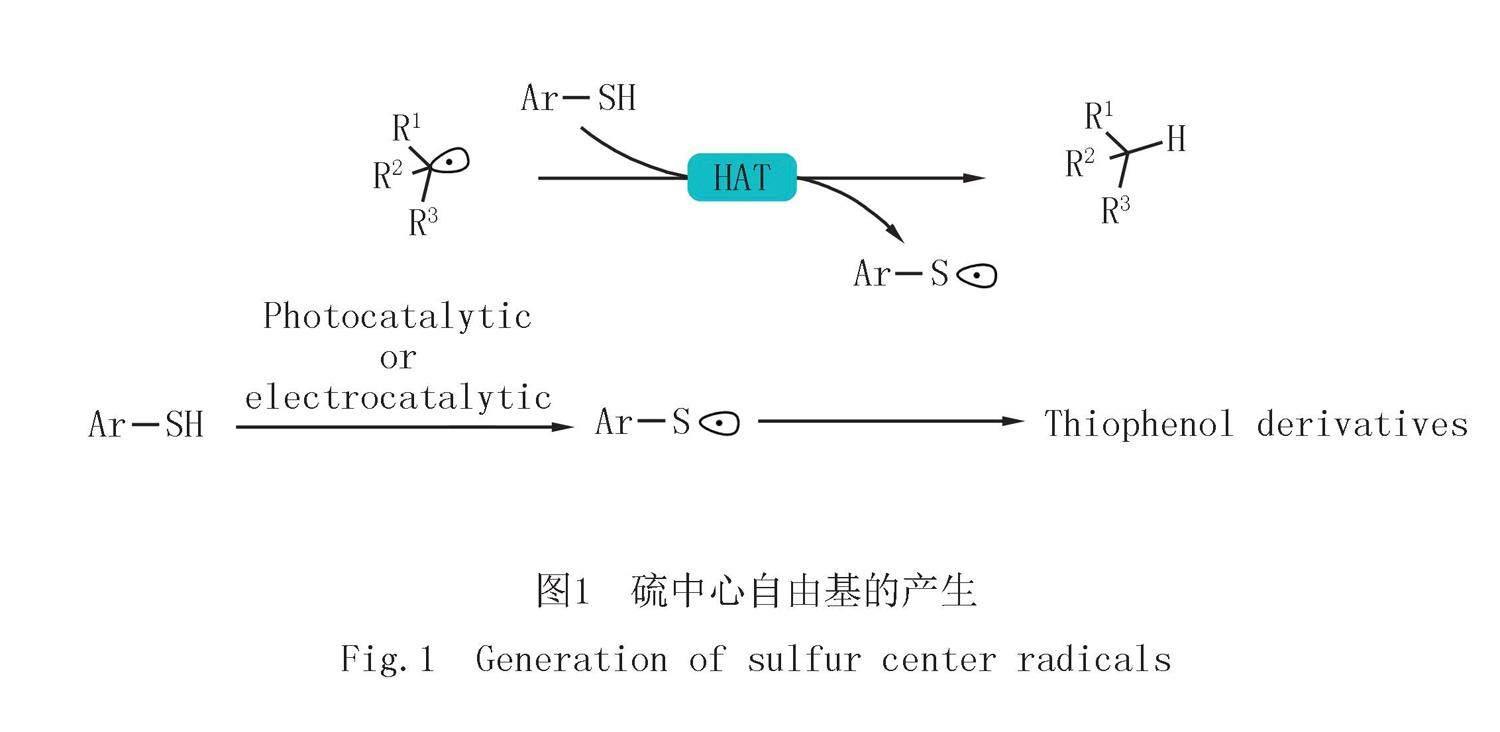
硫元素广泛存在于天然产物及药物分子中,构建含硫化合物在制药工业和合成化学上具有重要意义[1-2].化学家们发现通过硫酚产生的硫中心自由基可以作为高活性中间体,用于实现复杂含硫化合物的构建,如图1所示.硫鎓盐是最具活性的自由基前体之一,同时由于其正电性,硫鎓盐也是一类很好的电子受体.近年来硫鎓盐广泛用于EDA过程[3-5]中,具有很大的发展潜力[6-11].
黄原酸盐是在碱性条件下由醇对CS2亲核进攻生成的重要硫源试剂,在傅克反应[12]、三硫代碳酸酯的合成[13-14]、硫醇的合成[15]等过程中都有重要的应用.在有机全合成领域,黄原酸酯可以稳定活性自由基,从而提高活性自由基在高浓度介质中的存在时间,同时还可通过调节其绝对和相对浓度,使自由基加成到未活化的烯烃上[16].黄原酸酯还允许自由基添加到芳香环上,这一特性与烯烃的分子间加成结合,可用于构建3,4-二氢萘-1(2H)-酮化合物,在适当的条件下可以转化成萘.这种通用、高效的合成手段对于制药和农业化学工业以及材料科学来说非常重要[17-18].
由于黄原酸酯类化合物在药物化学、农业化学和材料科学等领域中的重要作用,近年来,开发更广泛的黄原酸酯类化合物引起了人们的广泛关注.S-芳基的黄原酸酯类化合物作为一类重要的黄原酸酯衍生物,其高效的合成策略一直存在一定的挑战.以往获得S-芳基黄原酸酯的方法都不太成功,存在硫醚作为唯一产物或副产物[19-20]的问题,如图2所示.最可靠的方法是基于1890年LEUCKART[21]提出的芳基重氮盐的芳基化.然而,由于芳基重氮盐存在着稳定性和安全性的问题,这种方法的应用受到限制.
随着可见光催化技术的发展,许多以前难以实现的反应可以在温和的条件下进行[22-26].光诱导的EDA机理由于其无金属性、温和性,最近也成为光催化合成领域的一种有效策略[27-30].本文报道了一种温和实用的方法,通过可见光激发噻蒽盐和黄原酸盐产生EDA复合物,从芳烃出发通过间接的C-H官能化策略实现芳基黄原酸酯类化合物的合成,如图3所示.
1 实验部分
1.1 仪器与试剂
所有溶剂与试剂(二异丙基乙胺,DIPEA;碳酸钠,Na2CO3;碳酸钾,K2CO3;叔丁醇锂,tBuOLi;三乙胺,Et3N;吡啶,Pyridine;二氯甲烷,DCM;N,N-二甲基甲酰胺,DMF;N-甲基吡咯烷酮,NMP;四氢呋喃,THF;1,4-二氧六环,1,4-Dioxane;二甲基亚砜,DMSO;乙腈,CH3CN;乙醇,EtOH;乙酸乙酯,EtOAc)均为分析纯,除特殊说明否则使用时未经进一步纯化.核磁共振测试中所用溶剂为CDCl3(氘代氯仿)或 DMSO-d6(氘代二甲基亚砜),1H NMR所用频率为500 MHz,13C NMR所用频率为125 MHz.“δ”表示化学位移,“J”表示耦合常数.所有反应通过用GF254硅胶涂覆的TLC(薄层色谱)板监测.文中所有化合物均是通过使用200~300目硅胶填充的高压快速柱色谱法分离纯化.
1.2 实验方法
首先向烘箱干燥过的25 mL Schlenk管中加入磁力搅拌子,然后依次加入噻蒽盐1a(0.3 mmol)、黄原酸盐2a(0.4 mmol).加入完毕之后将Schlenk管置换氮气至少3次.最后在氮气保护下使用注射器加入反应所需溶剂DCM 2 mL.随后封好管口,确保Schlenk管内体系被氮气保护并处于封闭状态,将Schlenk管置于蓝光灯处,在室温下进行光照并搅拌.反应一段时间后使用TLC技术对反应进行监测,直至反应结束.反应结束后,向体系中加入3~4 mL饱和食盐水,然后加入EtOAc进行萃取,得到的有机相进行合并,无水硫酸钠干燥后过滤并减压旋蒸.最后将得到的体系通过柱层析法对体系进行分离纯化,得到最终的产物.
1.3 产物表征数据
O-环己基-S-(3-甲酰基-4-甲氧基)苯基黄原酸酯(3a).产物性状:淡黄色液体;产率:85%(79 mg);洗脱剂:V(石油醚)/V(乙酸乙酯)= 20/1.1H NMR(500 MHz,CDCl3)δ: 10.45~10.43(m,1H),7.95(d,J=2.3 Hz,1H),7.65(dd,J=8.7,2.3 Hz,1H),7.05(d,J=8.7 Hz,1H),5.53~5.47(m,1H),3.98(s,3H),1.88~1.83(m,2H),1.57~1.51(m,4H),1.48~1.43(m,1H),1.39~1.33(m,2H),1.29~1.24(m,1H).13C NMR(CDCl3,125 MHz)δ: 212.10,188.64,162.68,142.47,135.69,125.34,122.40,112.55,82.97,56.01,30.56,25.13,23.10.
O-環戊基-S-(3-甲酰基-4-甲氧基)苯基黄原酸酯(3b).产物性状:淡蓝色液体;产率:85%(76 mg);洗脱剂:V(石油醚)/V(乙酸乙酯)=20/1.1H NMR(500 MHz,CDCl3)δ: 10.48(s,1H),7.96(d,J=2.4 Hz,1H),7.67(dd,J=8.7,2.4 Hz,1H),7.08(d,J=8.7 Hz,1H),5.84~5.79(m,1H),4.02(s,3H),1.94~1.88(m,2H),1.85~1.80(m,2H),1.63~1.59(m,4H).13C NMR(CDCl3,125 MHz,ppm)δ:212.14,188.65,162.66,142.42,135.66,125.32,122.47,112.51,88.14,56.00,32.46,23.74.
O-(4-苯丁基)-S-(3-甲酰基-4-甲氧基)苯基黄原酸酯(3c).产物性状:淡黄色液体;产率:72%(78 mg);洗脱剂:V(石油醚)/V(乙酸乙酯)=20/1.1H NMR(500 MHz,CDCl3)δ:10.42(s,1H),7.95(t,J=2.6 Hz,1H),7.65(dd,J=8.7,2.4 Hz,1H),7.28(t,J=7.5 Hz,2H),7.19(t,J=7.3 Hz,1H),7.12(d,J=7.2 Hz,2H),7.02(d,J=8.7 Hz,1H),4.55(t,J=6.4 Hz,2H),3.95(s,3H),2.57(t,J=7.7 Hz,2H),1.76~1.71(m,2H),1.63~1.56(m,2H).13C NMR(CDCl3,125 MHz)δ:213.14,188.56,162.76,142.60,141.77,135.73,128.36,128.32,125.89,125.42,122.18,112.64,74.35,55.98,35.29,27.72,27.53.
O-环己基-S-对甲苯基黄原酸酯(3d).产物性状:淡黄色液体;产率:82%(66 mg);洗脱剂:V(石油醚)/V(乙酸乙酯)=100/1.1H NMR(500 MHz,CDCl3)δ:7.38(d,J=8.0 Hz,2H),7.23(d,J=7.9 Hz,2H),5.56~5.49(m,1H),2.39(s,3H),1.88~1.79(m,2H),1.61~1.49(m,4H),1.39~1.23(m,4H).13C NMR(CDCl3,125 MHz)δ:212.78,140.16,134.89,129.87,126.79,82.42,30.48,25.13,22.94,21.38.
O-环戊基-S-对甲苯基黄原酸酯(3e).产物性状:淡黄色液体;产率:80%(61 mg);洗脱剂:V(石油醚)/V(乙酸乙酯)=100/1.1H NMR(500 MHz,CDCl3)δ:7.36(d,J=8.1 Hz,2H),7.22(d,J=7.9 Hz,2H),5.78~5.82(m,1H),2.39(s,3H),1.89~1.76(m,4H),1.59~1.55(m,4H).13C NMR(CDCl3,125 MHz)δ:212.80,140.14,134.84,129.85,126.87,87.66,32.38,23.65,21.37.
O-4-叔丁苯基-S-(对甲苯基)黄原酸酯(3f).产物性状:淡黄色液体;产率:70%(66 mg);洗脱剂:V(石油醚)/V(乙酸乙酯)=100/1.1H NMR(500 MHz,CDCl3)δ: 7.39(d,J=8.0 Hz,2H),7.30(t,J=7.5 Hz,2H),7.21(dd,J=12.3,7.7 Hz,3H),7.13(d,J=7.3 Hz,2H),4.56(t,J=6.3 Hz,2H),2.58(t,J=7.7 Hz,2H),2.39(s,3H),1.76~1.70(m,2H),1.62~1.56(m,2H).13C NMR(CDCl3,125 MHz)δ:213.72,141.79,140.32,134.97,129.98,128.34,128.29,126.60,125.82,73.99,35.25,27.69,27.47,21.37.
O-环己基-S-苯基黄原酸酯(3g).产物性状:淡黄色液体;产率:80%(61 mg);洗脱剂:V(石油醚)/V(乙酸乙酯)=100/1.1H NMR(500 MHz,CDCl3)δ:7.54~ 7.49(m,2H),7.47~7.39(m,3H),5.57~5.50(m,1H),1.86~1.77(m,2H),1.59~1.53(m,2H),1.46~1.40(m,2H),1.39~1.31(m,2H),1.30~1.23(m,1H).13C NMR(CDCl3,125 MHz)δ:211.93,134.97,130.09,129.85,129.09,82.39,30.41,25.11,22.82.
O-环戊基-S-苯基黄原酸酯(3h).产物性状:淡黄色液体;产率:75%(54 mg);洗脱剂:V(石油醚)/V(乙酸乙酯)=100/1.1H NMR(500 MHz,CDCl3)δ:7.52~7.46(m,2H),7.44~7.39(m,3H),5.82~5.77(m,1H),1.89~1.81(m,2H),1.78~1.76(m,2H),1.58~1.52(m,4H).13C NMR(CDCl3,125 MHz)δ:211.94,134.90,130.17,129.83,129.06,87.73,32.37,23.61.
O-環己基-S-(3,4-二甲氧基)苯基黄原酸酯(3i).产物性状:淡黄色液体;产率:79%(74 mg);洗脱剂:V(石油醚)/V(乙酸乙酯)=100/1.1H NMR(500 MHz,CDCl3)δ:7.07(d,J=8.3 Hz,1H),7.00(s,1H),6.89(d,J=8.3 Hz,1H),5.56~5.48(m,1H),3.91(s,3H),3.87(s,3H),1.82~1.81(m,2H),1.59~1.48(m,4H),1.43~1.26(m,4H).13C NMR(CDCl3,125 MHz)δ:212.98,150.59,149.08,128.12,121.32,117.92,111.28,82.35,56.01,55.90,30.51,25.12,22.93.
O-环己基-S-4-(叔丁基)苯基黄原酸酯(3j).产物性状:淡黄色液体;产率:81%(75 mg);洗脱剂:V(石油醚)/V(乙酸乙酯)=100/1.1H NMR(500 MHz,CDCl3)δ: 7.43(d,J=2.5 Hz,4H),5.56~5.53(m,1H),1.75(s,2H),1.59(d,J=6.4 Hz,2H),1.40~1.28(m,15H).13C NMR(CDCl3,125 MHz)δ:212.38,153.28,134.63,126.77,126.13,81.96,34.80,31.17,30.32,25.14,22.52.
O-环己基-S-4-(4-溴苯氧基)苯基黄原酸酯(3k).产物性状:淡黄色液体;产率:92%(117 mg);洗脱剂:V(石油醚)/V(乙酸乙酯)=90/1.1H NMR(500 MHz,CDCl3)δ:7.47(ddd,J=8.2,5.0,1.9 Hz,4H),7.01(dd,J=8.6,1.8 Hz,2H),6.93(dd,J=8.7,1.8 Hz,2H),5.59~5.51(m,1H),1.82(s,2H),1.62~1.55(m,2H),1.52~1.32(m,6H).13C NMR(CDCl3,125 MHz)δ:212.22,158.57,155.36,136.88,132.89,124.38,121.19,118.86,116.66,82.40,30.42,25.14,22.83.
O-环己基-S-4-(1,1'-联苯)基黄原酸酯(3l).产物性状:淡黄色液体;产率:86%(65 mg);洗脱剂:V(石油醚)/V(乙酸乙酯)=100/1.1H NMR(500 MHz,CDCl3)δ: 7.64(dd,J=14.2,7.9 Hz,4H),7.58(d,J=8.2 Hz,2H),7.48(t,J=7.6 Hz,2H),7.40(t,J=7.3 Hz,1H),5.60~5.53(m,1H),1.89~1.81(m,2H),1.63~1.57(m,2H),1.50~1.48(m,2H),1.46~1.40(m,1H),1.40~1.34(m,2H),1.31~1.25(m,1H).13C NMR(CDCl3,125 MHz)δ:211.88,142.67,139.95,135.27,128.91,128.87,127.88,127.71,127.13,82.49,30.44,25.11,22.86.
O-(4-叔丁氧基)-S-4-(1,1'-联苯)基黄原酸酯(3m).产物性状:淡黄色液体;产率:66%(75 mg);洗脱剂:V(石油醚)/V(乙酸乙酯)=100/1.1H NMR(500 MHz,CDCl3)δ:7.64(d,J=8.2 Hz,2H),7.59(dd,J=12.3,8.0 Hz,4H),7.48(t,J=7.5 Hz,2H),7.40(t,J=7.3 Hz,1H),7.23(t,J=7.3 Hz,2H),7.17(t,J=7.2 Hz,1H),7.10(d,J=7.3 Hz,2H),4.58(t,J=6.3 Hz,2H),2.56(t,J=7.7 Hz,2H),1.63~1.57(m,2H),1.63~1.57(m,2H).13C NMR(CDCl3,125 MHz)δ:212.87,142.80,141.72,139.84,135.35,128.88,128.67,128.28,127.93,127.80,127.15,125.81,74.09,35.26,27.70,27.54.
2 结果与讨论
2.1 反应条件的优化
以1a(芳基硫鎓盐)、2a(环己基黄原酸钠)作为反应的模板底物,对反应的最佳条件进行筛选.首先,在室温下用455 nm蓝光照射, DCM作为反应溶剂,对碱进行优化(表1,条目1~7),在使用Et3N作为碱时,反应产物的产率最高,达到了82%.同时实验结果表明在无碱的情况下反应产物的产率没有明显变化,说明碱在反应中对体系没有很大的促进作用(表1,条目8).然后对反应溶剂进行优化(表1,条目8~16),结果表明,CH3CN是反应的最佳溶剂.为了进一步提高转化率,使用了DMSO与H2O作为混合溶剂进行探索.实验结果表明,随着H2O在混合溶剂中比例的提高,产物的产率并没有提高,反而略微下降(表1,条目17~19),综上选用CH3CN作为体系的最佳溶剂.在确定了反应体系的溶剂之后,对光照条件进行了探索:在室温、黑暗环境中,并没有检测到反应产物,说明光照在反应过程中是必要的(表1,条目20).随后探索了温度对反应的影响,在避光的条件下,升高反应温度,结果表明,温度的升高会导致产率降低(表1,条目21~22).根据一系列的优化实验,得到反应的最佳条件为以乙腈作为反应溶剂,在室温及氮气保护下光照,反应时间为24 h(表1,条目14).
2.2 反应底物拓展
在确定最佳的反应条件后,为了探究反应策略对不同官能团的耐受性,对不同取代的硫鎓盐1和黄原酸盐2的底物适应范围进行了探究,结果总结在表2中.实验结果表明,不同的反应底物均能生成对应的S-芳基黄原酸酯3,并且产率良好(3a~3m).首先使用环己醇、环戊醇、1-苯基-4-丁醇合成对应的黄原酸盐与芳基硫鎓盐进行拓展.由邻甲氧基苯甲醛合成对应的硫鎓盐与黄原酸盐生成的衍生物3a~3c,产率在72%~85%.随后又尝试了给电子基团的芳基合成的硫鎓盐进行拓展.对甲基苯合成的硫鎓盐以70%~82%的产率得到产物3d~3f.而以苯合成的硫鎓盐与黄原酸盐反应生成的底物3g、3h也以80%、75%的产率得到.当以其他取代苯如邻二甲氧基苯、叔丁基苯也都分别以79%、81%的产率得到3i~3j.其他芳香化合物如4-溴联苯醚合成的硫鎓盐与环己基黄原酸盐也得到对应的衍生物3k,产率为92%;联苯合成的硫鎓盐也得到了对应的产物3l、3m,产率为86%、66%.上述结果表明,在此策略中各种芳基取代的硫鎓盐都表现出了良好的耐受性,证明了该策略具有良好的底物范围,对S-芳基黄原酸酯类化合物的研究起到重要的促进作用.
2.3 反应应用性研究
接下来,为了证明反应具有潜在的应用价值,以硫鎓盐1a、黄原酸盐2a为底物进行了克级转化,以78%的收率得到了目标产物3a.此外,为了证明体系对反应条件的耐受性,还进行了日光实验,在日光下以71%的产率得到产物3a(图4).这些结果充分证明了该策略潜在的价值.
此外,在标准条件下处理硫鎓盐1a与黄原酸盐2a反应的产物后,还以94%的分离产率回收了噻蒽,可以继续合成5-氧化噻蒽,进而合成硫鎓盐.这证明此策略良好的原子经济性,如图5所示.
2.4 反应机理研究
2.4.1 自由基捕获实验
为了进一步探究反应机理,进行了自由基捕获实验.首先,在标准条件下,在反应体系里加入了0.6 mmol TEMPO(四甲基哌啶氧化物,一种常见的自由基捕获剂).实验结果表明,3a的形成被完全抑制,如图6.这一实验结果证明了反应转化中涉及自由基的过程,但没有捕获到自由基中间体,可能是该反应体系中自由基物种过于活泼导致.
2.4.2 可能的机理
在初步实验结果和已有报道的基础上,提出了一种合理的反应机理.首先,硫鎓盐1与黄原酸盐2通过阴离子交换过程反应生成 EDA复合物A.随后EDA复合物A被光激发变为激发态B,并且在光诱导下进行从黄原酸盐阴离子到硫鎓盐1诱导的单电子转移(SET),产生一个自由基中间体C和一个硫中心自由基D.自由基C经历不可逆的裂解以产生芳基自由基F和可以循环利用的噻蒽E.最后,硫中心自由基D與芳基自由基F通过偶联以产生目标产物3.
3 结 论
综上所述,利用光激活EDA复合物,在温和无金属的条件下高效地实现了C-S键的构建,得到了一系列S-芳基黄原酸酯类化合物,并成功放大到克级,克服了这类化合物难以高效合成的难题.以噻蒽盐为芳基化试剂和电子受体,再一次证明了噻蒽盐应用的巨大潜力.此外,S-芳基黄原酸酯有望通过后期修饰,促进新的医用药物和农药的出现.
参 考 文 献
[1] FENG M H,TANG B Q,LIANG S H,et al.Sulfur containing scaffolds in drugs:synthesis and application in medicinal chemistry[J].Current Topics in Medicinal Chemistry,2016,16(11):1200-1216.
[2]ZHAO J Y,JIANG X F.The application of sulfur-containing peptides in drug discovery[J].Chinese Chemical Letters,2018,29(7):1079-1087.
[3]XU H,LI X F,DONG Y Z,et al.Thianthrenium-enabled phosphorylation of aryl C-H bonds via electron donor-acceptor complex photoactivation[J].Organic Letters,2023,25(20):3784-3789.
[4]XU H,LI X F,MA J,et al.An electron donor-acceptor photoactivation strategy for the synthesis of S-aryl dithiocarbamates using thianthrenium salts under mild aqueous micellar conditions[J].Chinese Chemical Letters,2023,34(11):108403.
[5]CABRERA-AFONSO M J,GRANADOS A,MOLANDER G A.Sustainable thioetherification via electron donor-acceptor photoactivation using thianthrenium salts[J].Angewandte Chemie(International Ed in English),2022,61(22):e202202706.
[6]BERGER F,PLUTSCHACK M B,RIEGGER J,et al.Site-selective and versatile aromatic C-H functionalization by thianthrenation[J].Nature,2019,567(7747):223-228.
[7]ENGL P S,HRING A P,BERGER F,et al.C-N cross-couplings for site-selective late-stage diversification via aryl sulfonium salts[J].Journal of the American Chemical Society,2019,141(34):13346-13351.
[8]LI J K,CHEN J T,SANG R C,et al.Photoredox catalysis with aryl sulfonium salts enables site-selective late-stage fluorination[J].Nature Chemistry,2020,12(1):56-62.
[9]YE F,BERGER F,JIA H,et al.Aryl sulfonium salts for site-selective late-stage trifluoromethylation[J].Angewandte Chemie(International Ed in English),2019,58(41):14615-14619.
[10]XU P,ZHAO D,BERGER F,et al.Site-selective late-stage aromatic[(18)F]fluorination via aryl sulfonium salts[J].Angewandte Chemie(International Ed in English),2020,59(5):1956-1960.
[11]SANG R C,KORKIS S E,SU W Q,et al.Site-selective C-H oxygenation via aryl sulfonium salts[J].Angewandte Chemie(International Ed in English),2019,58(45):16161-16166.
[12]KAWATA T,HARANO K,TAGUCHI T.Catalytic rearrangement of xanthates to dithiolcarbonates[J].Chemical and Pharmaceutical Bulletin,1973,21(3):604-608.
[13]YOSHIDA H,OGATA T,INOKAWA S.The conversion of some alkylxanthates to the corresponding trithiocarbonates.the nucleophilic reaction of -SCSSR[J].Bulletin of the Chemical Society of Japan,1971,44(11):3106-3108.
[14]OVERBERGER C G,DRUCKER A.The preparation of meso-and DL-α,α'-stilbenedithiol,meso-and DL-2,3-butanedithiol,and DL-1,2,3,4-butanetetrathiol[J].The Journal of Organic Chemistry,1964,29(2):360-366.
[15]DJERASSI C,GORMAN M,MARKLEY F X,et al.Studies in organic sulfur compounds.VII.1 lithium aluminum hydride reduction of xanthates to mercaptans.synthesis of substituted β-mercaptoethanols[J].Journal of the American Chemical Society,1955,77(3):568-571.
[16]QUICLET-SIRE B,ZARD S Z.On the strategic impact of the degenerative transfer of xanthates on synthetic planning[J].Israel Journal of Chemistry,2017,57(3/4):202-217.
[17]ZHAO B W,LI J J,PAN X Q,et al.Photoinduced free radical promoted cationic RAFT polymerization toward "living" 3D printing[J].ACS Macro Letters,2021,10(10):1315-1320.
[18]BARTHET C,WILSON J,CADIX A,et al.Micellar RAFT/MADIX polymerization[J].ACS Macro Letters,2017,6(12):1342-1346.
[19]VOLKOV A A,BUGAENKO D I,BOGDANOV A V,et al.Visible-light-driven thioesterification of aryl halides with potassium thiocarboxylates:transition-metal catalyst-free incorporation of sulfur functionalities into an aromatic ring[J].The Journal of Organic Chemistry,2022,87(12):8170-8182.
[20]HE R F,LIU Y,FENG Y Q,et al.Access to thienopyridine and thienoquinoline derivatives via site-selective C-H bond functionalization and annulation[J].Organic Letters,2022,24(17):3167-3172.
[21]LEUCKART R.Eine neue methode zur darstellung aromatischer mercaptane[J].Journal Für Praktische Chemie,1890,41(1):179-224.
[22]YU X Y,CHEN J R,XIAO W J.Visible light-driven radical-mediated C-C bond cleavage/functionalization in organic synthesis[J].Chemical Reviews,2021,121(1):506-561.
[23]GENTRY E C,KNOWLES R R.Synthetic applications of proton-coupled electron transfer[J].Accounts of Chemical Research,2016,49(8):1546-1556.
[24]ARORA A,WEAVER J D.Visible light photocatalysis for the generation and use of reactive azolyl and polyfluoroaryl intermediates[J].Accounts of Chemical Research,2016,49(10):2273-2283.
[25]ENGL S,REISER O.Copper-photocatalyzed ATRA reactions:concepts,applications,and opportunities[J].Chemical Society Reviews,2022,51(13):5287-5299.
[26]CHANG L,AN Q,DUAN L F,et al.Alkoxy radicals see the light:new paradigms of photochemical synthesis[J].Chemical Reviews,2022,122(2):2429-2486.
[27]CHEN Z R,XUE F,LIU T X,et al.Synthesis of β-hydroxysulfides via visible-light-driven and EDA complex-promoted hydroxysulfenylation of styrenes with heterocyclic thiols in EtOH under photocatalyst-free conditions[J].Green Chemistry,2022,24(8):3250-3256.
[28]WOZ′NIAK ,MURPHY J J,MELCHIORRE P.Photo-organocatalytic enantioselective perfluoroalkylation of β-ketoesters[J].Journal of the American Chemical Society,2015,137(17):5678-5681.
[29]LIANG X,LI Y F,XIA Q,et al.Visible-light-driven electron donor-acceptor complex induced sulfonylation of diazonium salts with sulfinates[J].Green Chemistry,2021,23(22):8865-8870.
[30]YANG Z L,LIU Y T,CAO K,et al.Synthetic reactions driven by electron-donor-acceptor(EDA) complexes[J].Beilstein Journal of Organic Chemistry,2021,17:771-799.
A new strategy for the synthesis of S-arylxanthate via electron donor acceptor complex photoactivation
Yang Daoshan, Zuo Junze
(College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, China)
Abstract: Xanthate esters have been attacting chemists' interest as an important component in medicinal chemistry, agricultural chemistry, and materials chemistry. However, the efficient synthesis of S-aryl-substituted xanthate esters has still faced challenges. Herein, we report a new strategy for the construction of C-S bonds based on the EDA(electron donor acceptor) process under light-induced conditions. A series of S-aryl xanthates were obtained in moderate to excellent yields under mild metal-free conditions. Gram-scale and sunlight experiments demonstrated the potential applications of the reaction, which will provide a new strategy for the synthesis of xanthate esters.
Keywords: xanthate esters; electron donor acceptor complex; thianthrenium salts; visible light induction; construction of C-S bond
[責任编校 赵晓华 陈留院]
