基于PTP1B 活性区域靶向中药成分治疗糖尿病的虚拟筛选及动力学模拟验证
2024-05-14杨子博支爽代霖霖王慧李冬冬
杨子博,支爽,代霖霖,王慧,李冬冬
天津市医药科学研究所,天津 300020
2 型糖尿病发病机制复杂,很难找到治疗2 型糖尿病的完美机制。最近,治疗糖尿病最常用的药物二甲双胍被发现含有致癌结构N-亚硝基二甲胺[1]。新的药物并没有彻底缓解全球糖尿病危机,持续增加的糖尿病患者依旧需要新的药物。天然产物的半合成是一种更有效、更安全的药物开发策略,对于改进药物特性、增强药动学和药效学至关重要[2]。本研究尝试利用计算机辅助技术在中药成分中筛选治疗糖尿病的化学结构。
蛋白酪氨酸磷酸酶1B(PTP1B)是胰岛素信号通路的负调节因子,在与胰岛抵抗相关的疾病中起着非常关键的作用[3]。清除神经元或全身的PTP1B,小鼠可以有效的免于肥胖和糖尿病[4]。由于PTP1B在血管内皮生长因子(VEGF)通路中负调节,抑制PTP1B 也可以促进心血管再生[3]。PTP1B 还与β 细胞增生相关,敲除掉小鼠的β 细胞后,小鼠获得了更高水平的β 细胞增生及葡萄糖引起的胰岛素分泌[1]。由于PTPs 结构的相似性,PTP1B 抑制剂缺乏特异性,部分药物如trodusquemine、JTT-551 等的临床试验由于缺乏特异性并且存在不良反应而被中止[5]。
PTP1B 有2 个活性区域[6]。PTP1B 抑制剂设计常用的活性区域是由编号为210~223 的残基组成的区域A。多数PTP1B 抑制剂是针对区域A 的,但是单独针对区域A 的结构选择性较差[5]。由于区域A 呈现强极性,针对区域A 设计的配体膜透过性比较差。为了提高结构特异性,还要考虑另外3 个区域(区域B、C、D)的共同作用。区域B 由残基Tyr-20、Arg-24、Ala-27、Phe-52、Arg-254、Met-258、Gly-259 组成,同时作用于区域A 和区域B 的结构有更好的选择性[7]。区域C 是主要由46~48 号残基组成的带电区域,可以形成盐桥、氢键等,在选择性设计上使用更多。D 区域由Glu-115、Lys-120、Asp-181、Ser-216 组成,该区域选择性更强,Lys-120在区分 TPT1B 和 T 细胞蛋白酪氨酸磷酸酶(TCPTP)时起决定作用。多羟基或羧基的抑制剂可以提高20 倍与Lys 的结合能力[8]。B 区域的Arg-24、Arg-254 和D 区域的Lys-120 是关键残基。综上,本研究针对PTP1B 活性区域A 及附近可以提高结构特异性区域,利用TCMSP 数据库[9]进行PTP1B 潜在抑制剂结构筛选。
1 方法
1.1 对接筛选
从PDB 数据库[10]下载蛋白3D 结构(PDB ID:2BGE),检查、修复结构文件,利用Autodock Tools生成PDBQT 格式文件。设定在A、B、C、D 4 个区域内为对接口袋,对接盒子参数为--center_x56.9--center_y35.4--center_z40.3--size_x32.1--size_y36.7--size_z28.8。利用OpenBabel 生成TCMSP 数据库的中药成分分子性质,筛选相对分子质量<500,氢键供体≤5,氢键受体≤10,lgP≤5 的结构,利用Autodock Vina 做筛选,按对接能绝对值由大到小取前100 个结构。利用LeDock 做筛选,按对接能绝对值由大到小取前100 个结构。对2 次对接结果取交集,利用 Pymol、Discovery Studio Visualiser(DSV)、LigPlot+[11]分析与活性区域A 存在作用且同时与B、C 或D 区域存在作用的中药成分。
1.2 结构分析
利用Openbabel[12]工具生成结构SMILS 信息,在 SwissADME[13]中计算拓扑分子极性表面积(TPSA)、lgP、氢键供体、氢键受体等信息,筛选TPSA≤140 Å2(1 Å=0.1 nm),lgP≤5,氢键供体≤5,氢键受体≤10,S.A score<10 的结构,并判断结构是否满足Lipinski、Veber、Egan 成药原则。利用已知活性结构数据库PASS[14]分析结构潜在用途,筛选有活性的概率(Pa)>没有活性的概率(Pi)且与糖尿病相关的项目。
1.3 药动学预测
利用Openbabel等工具生成结构SMILES信息,在pkCSM 数据库[15]中计算结构药动学性质,统计结果并分析。稳态分布体积(VDss)>0.45 被认定为比较高的值;血脑屏障透过率(lgBB)>0.3 有较好的血脑屏障透过能力,lgBB<-1 则比较差;中枢神经系统通透性(lgPS)>-2 被认为可以进入中枢神经系统,lgPS<-3 则无法进入中枢神经系统;细胞色素P450(CYP)是一种重要的单氧酶,在组织中广泛存在,与药物代谢相关,CYP 对药物有抑制或激活作用,酶的抑制剂会破坏药物的代谢,可能产生与预期相反的效果。2D6 和3A4 是与代谢相关的CYP 2 个重要异构体。较低的清除率可以使药物被清除前更多到达治疗部位,Total clearance<0.5 被认为有较低的清除率。Ames 测试阴性说明结构无致癌风险;大鼠口服急性毒性(LD50)>2.0 被认为致癌风险高[16]。
1.4 分子动力学模拟验证
利用Gromacs 2019,使用AMBER99SB-ILDN力场,TIP3P 水模型,设置nstlist 为1、cutoff-sheme为Verlet、ns_type 为grid、rlist 为1.2 做能量最小化。设置continuation 为no、constraint_algorithm 为lincs、cutoff-scheme 为Verlet、coulombtype 为PME、tcoupl 为V-rescale 做NVT 平衡。设置pcoupl 为Berendsen、pcoupltype 为isotropic、tau_p 为2.0、ref_p 为1.0 做NPT 平衡,待体系平衡后做动力学模拟并以分子力学/广义波恩模型表面积(MM/GBSA)[17]分析主要作用力。
2 结果
2.1 对接筛选
根据分子性质筛选出2 857 个中药成分,利用Autodock Vina 筛选,如图1 所示,对接能最低的结构是双靛蓝(bisindigotin,MOL011100)和二氢血根碱(dihydrosanguinarine,MOL001463),对接能为-9.0 kcal/mol(1 cal=4.2 J);对接能最高的结构是五味子醇甲(schisandrin,MOL005604),对接能为-4.7 kcal/mol。Ledock 筛选结果中对接能最低的结构是taurochenideixycholicacid(MOL009666),对接能为-6.65 kcal/mol;对接能最高的结构是白前苷B(glaucoside B,MOL012930),对接能为-1.69 kcal/mol。Vina 给出的结合能更低。
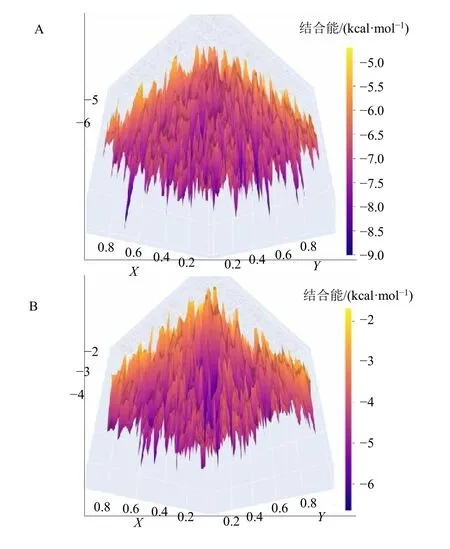
图1 Autodock Vina(A)和Ledock(B)对接结果统计Fig.1 Results of docking with Autodock Vina (A) and Ledock (B)
合并2 次筛选结果得到结构2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5Hbenzocycloheptene(MOL000225)、刺桐苯乙烯(eryvariestyrene,MOL000452)、掌叶大黄二蒽酮C(palmidin C,MOL002253)、5,7-dihydroxy-2-[(2S,3R)-2-(4-hydroxy-3-methoxyphenyl)-3-(hydroxymethyl)-2,3-dihydro-1,4-benzodioxin-6-yl]chromen-4-one(MOL003037)、甘草宁D(gancaonin D,MOL004861)、槲皮素-7-O-葡萄糖苷(quercetin 7-O-glucoside,MOL005921)、isoprincepin(MOL009551)、bisindigotin(MOL011100)、异丙甲素A(isatisine A,MOL011108)、异靛蓝(isoindigo,MOL011335),见表1。

表1 Autodock Vina 及Ledock 对接结果交集部分的结构Table 1 Structures from section of Autodock Vina and Ledock intersection
Pymol中显示2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 与残基Gln-266、Arg-221、Asp-181、Lys-120 形成氢键(图2A),在LigPlot+中显示2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5Hbenzocycloheptene 与残基Asp-48、Asp-181、Lys-120形成氢键,与残基Arg-221 形成2 个氢键,与亲水残基Tyr-46、Gln-262、Ser-216、Cys-215 存在非键作用,与疏水残基Ala-217、Gly-220、Phe182 存在非键作用(图2B)。

图2 2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 与PTP1B 对接结果在Pymol(A)和LigPlot+(B)中的分析Fig.2 Analysis of docking results of 2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene and PTP1B in Pymol (A) and LigPlot+(B)
2.2 结构分析
2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene、甘草宁D、槲皮素-7-O-葡萄糖苷、双靛蓝、异丙甲素A、异靛蓝满足所有类药原则。除结构掌叶大黄二蒽酮C和5,7-dihydroxy-2-[(2S,3R)-2-(4-hydroxy-3-methoxyphenyl)-3-(hydroxymethyl)-2,3-dihydro-1,4-benzodioxin-6-yl]chromen-4-one 外都有较高的吸收能力(ABS)。所有结构的S.A score<6,生物利用度为55%,见表2。

表2 类药性质分析Table 2 Drug-like properties analysis
PASS 数据库分析显示,除结构isoprincepin 和异丙甲素A外,其他结构都可以用于糖尿病的治疗,如antidiabetic(type 1)、antidiabetic、antidiabetic(type 2)。或者可以作用于胰岛素,如用于抑制insulysin的insulysin inhibitor,或增加胰岛素效用的insulin sensitizer、insulin promoter。此外,还有多个与葡萄糖相关的作用,如抑制碳水化合物水解的alpha glucosidase inhibitor、beta-glucosidase inhibitor,抑制半乳糖向葡萄糖转化的UDP-glucose 4-epimerase inhibitor 等与糖尿病相关的潜在作用,见表3。

表3 PASS 数据库分析结果Table 3 Results of PASS analysis
2.3 药动学预测
所有结构肠吸收能力都比较强,异丙甲素A 肠吸收比值相对低一些[15]。2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocyclo heptene 分布容积较高[18]。所有结构的血脑屏障通透性比较差,刺桐苯乙烯、双靛蓝、异靛蓝有可能可以进入中枢神经系统[15]。双靛蓝可以作为2D6 的底物,除 5,7-dihydroxy-2-[(2S,3R)-2-(4-hydroxy-3-methoxyphenyl)-3-(hydroxymethyl)-2,3-dihydro-1,4-benzodioxin-6-yl]chromen-4-one、槲皮素-7-O-葡萄糖苷、异丙甲素A 外都可以用作3A4 的底物。所有结构都不是2D6 的抑制剂,2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene等结构不是3A4 的抑制剂。所有结构都满足清除指数要求,且无致癌风险。LD50在2.069~2.740 mol/kg,说明这些结构只有在高剂量时才会致死,见表4。
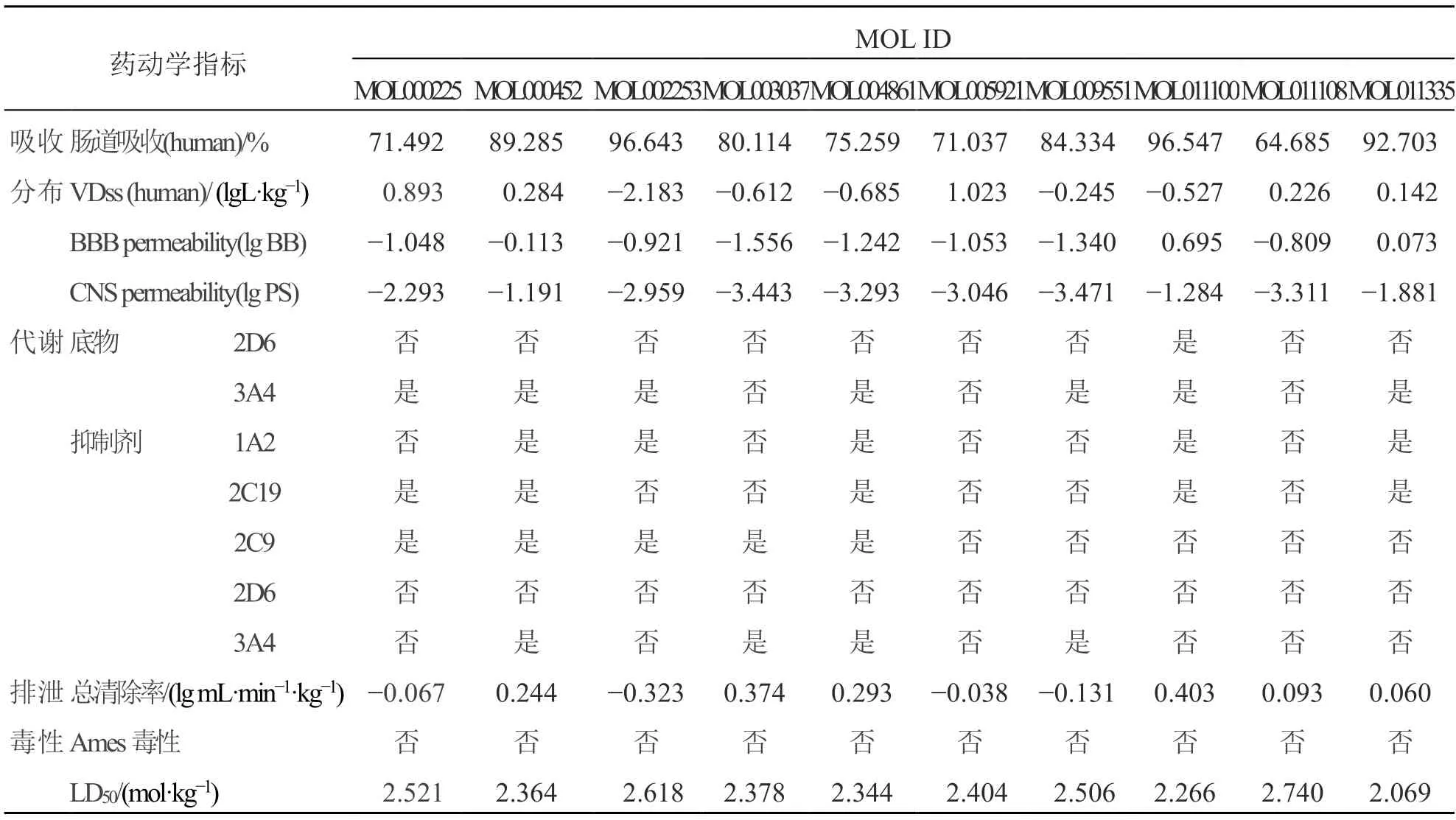
表4 药动学分析结果Table 4 Results of pharmacokinetic analysis
2.4 动力学模拟
PyMol分析显示,2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 与残基Gln-262、Gln-266、Arg-221、Asp-181、Asp-48 形成氢键(图3A)。LigPlot+分析显示,2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5Hbenzocycloheptene 与Asp-48、Asp-181、Gln-266、Arg-221 形成氢键,与疏水残基Phe-182、Gly-220、Ile-219,与亲水残基Gly-262,及残基Lys-120 存在非键作用(图3B)。2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 在动力学模拟中没有发生太大的构象变化(图3C)。DSV 分析显示,2,3,7-trihydroxy-5-(3,4-dihydroxy-Estyryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 与ASP-181、ASP-48、GLN-262 形成常规氢键,与Gly-220、Asp-48、Lys-120 形成碳氢键,与Asp-181 形成Pi-Donor 氢键,与Tyr-46、Ile-219 存在Pi/Alkyl疏水作用。羟基与亲水口袋的作用及七元环、苯环与疏水区域的作用使2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocy cloheptene稳定结合在活性区域和PTP1B 特征区域(图3D)。

图3 分子动力学模拟最后一帧中2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 构象与PTP1B 作用分析Fig.3 Analysis of the interaction between 2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5Hbenzocycloheptene and PTP1B in the last frame of molecular dynamics simulation
对 Backbone 做最小二乘拟合,分别计算Backbone 和2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 的均方根误差(RMSD),计算结果显示蛋白和2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocy cloheptene 在模拟中都非常稳定,RMSD 波动范围小于1 Å,见图4。
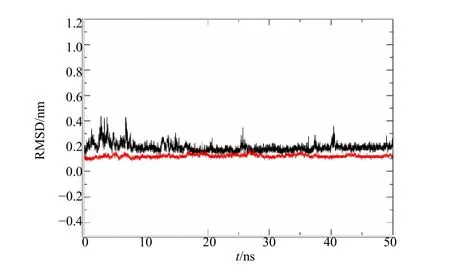
图4 2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 与PTP1B 结合的复合物RMSD 分析Fig.4 RMSD analysis of complex of 2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5Hbenzocycloheptene and PTP1B
分子动力学模拟轨迹氢键分析显示2,3,7-trihy droxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5Hbenzocycloheptene 与PTP1B 可以形成4 个左右氢键,键长约3 Å,见图5。

图5 2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 与PTP1B 氢键个数(A)及键长(B)Fig.5 Statistics of the number (A) and bond length (B) of hydrogen bonds between 2,3,7-trihydroxy-5-(3,4-dihydroxy-Estyryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene and PTP1B
相比关键残基Arg-24、Arg-254,2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5Hbenzocycloheptene 与关键残基Lys-120 发生作用的可能性更大,最近距离约为4 Å,可以形成1 个氢键,约50 个PTP1B 原子位于距离2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-ben zocycloheptene 5 Å 范围内,见图6。

图6 2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 与残基Lys-120 氢键个数(A)、键长(B)及5 Å 范围内PTP1B 原子数(C)Fig.6 Statistics of hydrogen bonds between 2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5Hbenzocycloheptene and residue Lys-120 (A),bond length (B),and statistics of PTP1B atoms within 5 Å (C)
2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 与PTP1B 复合物的范德华力贡献值为-29.59 kcal/mol,电子效应贡献值为-6.80 kcal/mol,极性作用在溶剂化自由能中的贡献值为8.50 kcal/mol,非极性在溶剂化自由能中的贡献值为-4.92 kcal/mol,气态作用能ΔGGAS为-36.39 kcal/mol,溶剂作用能ΔGSOLV 为-36.39 kcal/mol,总能量ΔTOTAL 为-32.82 kcal/mol,结合自由能ΔG 为-24.76 kcal/mol,见图7、表5。

表5 2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 与PTP1B 作用力Table 5 List of forces between 2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5Hbenzocycloheptene and PTP1B

图7 2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 与PTP1B 作用力类型分析Fig.7 Analysis of force types between 2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5Hbenzocycloheptene and PTP1B
3 讨论
Autodock Vina 和LeDock 对接结合能都比较低的结构有10个,其中2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 与PTP1B 的 Autodock Vina 对接结合能为-7.7 kcal/mol,说明其与PTP1B 结合比较稳定。与常用PTP1B 抑制剂靶向区域A 的残基Arg-221 形成2 个氢键,与PTP1B 选择性设计区域C 的残基Asp-48形成氢键,与选择性区域D 的残基Asp-181 及关键残基Lys-120 形成氢键,符合跨活性区域共同作用提高PTP1B 抑制剂选择性的设计要求。
2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 在胃肠道高吸收,TPSA 为101.1 Å2,可以通过细胞膜。lgP值1.67,说明其有良好的口服吸收,同时满足Lipinski[19]、Veber[20]、Egan[21]等的类药原则,合成难度较低。药物作用预测显示2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5Hbenzocycloheptene 可以用于治疗1 型糖尿病,或用作胰岛素激动剂。在pkCSM 数据库预测结果中表现出较好的药动学性质,肠吸收率71.492%,清除指数较低,能有效抵达治疗部位。Ames 测试阴性,无致癌风险。LD50值2.521 mol/kg,在较高剂量时才会致死[22]。
在分子动力学模拟0~1 ns 的区间内,2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 有小幅的构象变化,之后虽然有波动,但是根据RMSD 可以判断2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-ben zocycloheptene 能够以一个稳定构象稳定结合。对比受体RMSD 轨迹,受体在0~1 ns 区间内也有小幅构象转变,由此推测 2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocy cloheptene 在模拟开始的RMSD 变化,一定程度上可能是受体构象改变所影响。2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene
的稳定构象与A 区域残基Arg-221 和WPD 区域残基Asp-181 同时形成氢键。特征区域C 平而宽,完全暴露在蛋白表面。2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 与常见的PTP1B 抑制剂相同,在对接构象和动力学模拟构象中与区域C 的残基Asp-48 形成氢键。
气相自由能包括成键作用和非键作用,2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene与PTP1B 的结合不涉及成键作用,非键作用中范德华力起主要作用。溶剂化自由能包括极性作用和非极性作用,非极性作用值为-4.92 kcal/mol,占主导地位。2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 结合自由能为-24.76 kcal/mol,结合MM/GBSA 分析数值,说明2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 与PTP1B 的结合是一个范德华力主导的自发过程。
在TCMSP 数据库中显示2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5Hbenzocycloheptene 与豆蔻相关,豆蔻与糖尿病、肥胖、胰岛素依赖型糖尿病相关。2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5Hbenzocycloheptene 在PubChem 数据库中ID 是162880943,但是没有相关生物信息的记录,在Reaxys 数据库及常用搜索引擎中只有少量的相关信息。2001 年Kikuzaki 等[22]报道利用光谱法从大豆蔻中识别出2,3,7-trihydroxy-5-(3,4-dihydroxy-Estyryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene。
综上所述,豆蔻中的成分2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5Hbenzocycloheptene 在计算机辅助药物设计分析中表现出良好的类药性质及药动学性质,存在治疗糖尿病的潜在可能性。分子动力学模拟结果显示2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene 可以自发的与PTP1B 多个活性区域共同作用稳定结合,可能是一个PTP1B 的潜在特异性抑制剂,可以对2,3,7-trihydroxy-5-(3,4-dihydroxy-E-styryl)-6,7,8,9-tetrahydro-5H-benzocycloheptene
做进一步的结构优化及实验验证。
利益冲突所有作者均声明不存在利益冲突
