金属有机框架纳米材料在PET成像和肿瘤治疗中的应用研究
2024-05-13魏柯君薛长国
魏柯君,薛长国
1. 安徽理工大学 材料科学与工程学院,安徽 淮南 232001;2. 安徽医科大学附属巢湖医院 物资设备科,安徽 合肥 238000
引言
正电子发射计算机断层显像(Positron Emission Tomography,PET)是一种核医学成像技术,作为非侵入性的成像方式,具有更高的检测灵敏度(低至皮摩尔范围)、更深的信号穿透力以及更好的定量能力,从而在临床前和临床场景中得到更广泛的应用[1]。相较于其他成像技术,PET 成像专注体内的代谢过程和分子活动,从而使疾病得以在早期阶段被发现。金属有机框架(Metal-Organic Frameworks,MOF)也称为多孔配位聚合物,已成为生物医学领域最具吸引力的应用材料之一[2]。作为一类新兴的多功能材料,MOF 是由有机连接体(通过配位键)桥接的金属离子簇构成的。相较于传统纳米材料,纳米级金属有机框架(Nanoscale Metal-Organic Frameworks,nMOF)具有多重优势:优异的结构和组分可调性(允许生产具有不同形状、尺寸和化学性质的nMOF)、更具适应性的降解能力和更好的化学改性能力,这些显著优点使得nMOF 被用作癌症治疗工具,并装载各种治疗药物和成像材料[3-4]。
在癌症治疗管理模式快速发展的时代,科研人员对癌症生物学基本驱动因素的理解不断加深,肿瘤微环境的重要性,尤其是新型的生物化学材料和人体免疫系统的发展对癌症发生的作用愈发凸显。在我国科研人员对新型材料在癌症中作用研究不断深入的背景下,高效的纳米材料和靶向治疗策略已在临床试验阶段取得显著成果。在这些更为科学的治疗方法迅速发展的同时,新型纳米材料PET 示踪剂的成像和治疗能力已逐渐崭露头角,成为一个极具潜力的研究和发展方向。这将为肿瘤治疗提供一种创新策略,为患者带来更有效的治疗选择。
1 资料与方法
1.1 一般资料
有大量研究涉及利用nMOF 材料作为体内癌症治疗剂。例如,基于铪-二氢卟酚的nMOF 具有良好的结肠癌光动力疗法(Photodynamic Therapy,PDT)功效[5]。有研究报道,基于铪-四氢化葵(4-羧苯基)卟啉的nMOF 聚乙二醇(Polyethylene Glycol,PEG)功能化后,成功用于小鼠乳腺癌模型中的组合PDT 和放射治疗[6]。nMOF 卓越的载货能力使其适用于多种癌症类型的联合治疗,如小干扰RNA(siRNA)与化疗药物、双重化疗药物以及化疗药物与PDT 等不同治疗组合已在体内肿瘤治疗中得到尝试,并取得了鼓舞人心的成果[7-9]。另一方面,尽管已经收集到令人信服的证据证明nMOF 可以很容易地用于多种成像技术,如CT、MRI 和光学成像。但迄今为止,使用nMOF 作为体内肿瘤显像剂进行的研究非常有限[10-11]。
本研究的目标是表征适用于PET 成像和肿瘤靶向的生物相容性nMOF,为未来PET 引导下癌症治疗中的货物运输做好准备。基于最佳表面积比同时具有不依赖于连接剂的特殊稳定性,选择含锆的UiO-66 nMOF[以1,4-苯二甲酸酯和苯甲酸作为桥联基]作为模板材料[12-13]。此外,由于存在Zr6O4(OH)4连接簇[14],PET 同位素锆-89(89Zr,t1/2=78.4 h)可以在合成过程中无缝整合到UiO-66 结构中[15]。使用芘衍生的聚乙二醇(Pyrene-Derived Polyethylene Glycol,Py-PGA-PEG)进行表面工程[16]不仅可以提高UiO-66 在生物介质中的稳定性和分散性,而且还可以为肿瘤靶向分子的整合提供进一步的功能化位点[17]。同时,UiO-66 的高度多孔结构能够很好地容纳亲水性和疏水性药物,并且可以揭示其持续(长达30 d)的pH 依赖性药物释放曲线[18]。
1.2 nMOF的合成
通用的合成nMOF 的方法包括纳米沉淀法、溶剂热法、反相微乳液法和表面活性剂模板溶剂热反应法4 种。纳米沉淀法倾向于产生无定形材料;其他3 种方法可以提供结晶材料,能够更好地控制纳米粒子的成核和生长动力学。前两种方法不含表面活性剂;而后两种方法依赖表面活性剂,不仅可以控制颗粒合成,还可以稳定这些颗粒。
由于F3 肽可在肿瘤脉管系统中与肿瘤细胞和活化内皮细胞实现有效结合,故本研究选择其为肿瘤靶向配体[19]。先前的一项研究表明,F3 肽选择性地与肿瘤细胞表面的核仁素结合,随后可以转运到细胞核和细胞质中[20]。细胞表面核仁素表达升高是各种癌细胞,尤其是乳腺癌细胞的重要特征。将半胱氨酸残基掺入F3 肽的C 末端以促进其与UiO-66 nMOF 的缀合(通过硫醇马来酰亚胺与芘-PEG-马来酰亚胺反应)[21]。
在将F3 肽附着到UiO-66 上(最终缀合物将命名为UiO-66/Py-PGA-PEG-F3)后,进行体外测定(如采用流式细胞术和共聚焦荧光显微镜)来验证UiO-66/Py-PGAPEG-F3 对细胞核仁素的靶向特异性。随后在携带原位MDA-MB-231 肿瘤的小鼠身上进行体内PET 成像、器官分布和组织学研究,以确认UiO-66/Py-PGA-PEG-F3的肿瘤靶向能力。阿霉素(Doxorubicin,DOX)加载到UiO-66 结合物上,作为模型抗癌药物和荧光团来定义纳米结合物的位置。除此之外,通过药物递送研究进行体内原理验证,以探究静脉注射载有DOX 的UiO-66 偶联物后DOX 增强的肿瘤靶向递送效率,合成过程如图1 所示。经Py-PGA-PEG 适度功能化后的UiO-66 偶联物在小鼠体内表现出良好的生物相容性,组织学染色和血清生化测定的分析结果证实其不会导致急性或慢性毒性。由此推断,本质上具有放射性的UiO-66 结合物显示出巨大的应用潜力,未来可用于图像引导的药物递送治疗和靶向癌症治疗。

图1 89Zr-UiO-66/Py-PGA-PEG-F3偶联物合成路线
1.2.1 在nMOF中整合生物相容性抗癌药物
现阶段可以利用nMOF 的可定制性以提供高药剂负载,将成像和治疗剂纳入nMOF。这些加载方法分为两大类:在nMOF 合成期间直接掺入或合成后加载。这两种策略的组合也可用于将不同的试剂加载到nMOF 中,以开发多模式成像剂或治疗诊断纳米颗粒。
在合成后加载策略中,生物医学相关试剂被加载到nMOF 孔中。在高度多孔的nMOF 合成后,活性剂通过非共价或共价相互作用结合到nMOF 中,首次使用体相MOF 证明了非共价药物负载。释放研究表明,药物从框架中释放非常缓慢、持续,且爆发效应极小。相关研究已将该策略扩展到负载有亲水性、两亲性和疏水性药物的nMOF,nMOF 孔径必须大于封装剂才能承受高负载[22]。
由于非共价药物加载是一个固有的可逆过程,因此nMOF 的后续处理可能会导致药物过早释放。药物合成后的共价附着提供了一种更可靠的方法,因为药物只有在NMOF 分解时才会被释放。在这种方法中,多孔nMOF 框架内的正交官能团用于共价连接活性剂。该策略有效地创建了前药,因此必须保持代理的功能。在特定的生物条件下,活性药物必须可从nMOF 上裂解。由于纳米粒子表面或附近的官能团在动力学上更容易接近,因此整个nMOF 粒子的药剂负载可能不均匀。
1.2.2 pH值对抗癌药物释放的影响
UiO-66 nMOF 具有相对较高的表面积比,通过氮气吸附-脱附等温线测量的结果(988.625 m2/g)证实其适合装载各种货物。Py-PGA-PEG-F3 功能化后表面积降至438.978 m2/g,如图2 所示根据DOX 在488 nm 处的吸光度测量,计算出可加载到UiO-66 中的DOX 量为1 mg DOX/mg UiO-66。DOX@UiO-66/Py-PGA-PEG-F3 的DOX 释放曲线在模拟生理条件下进行测试,pH 值分别为5.0、6.8和7.4,温度为37℃。如图3 所示,中等pH 值对UiO-66 偶联物中DOX 的释放速率有影响。在pH 值为7.4 时,2 周后可以释放27.73% 的DOX(0.27 mg DOX/mg UiO-66),这表明UiO-66 结构中负载的DOX 在生理条件下相对稳定。当暴露于模拟肿瘤细胞外pH 值为6.8 时,DOX 的释放百分比在同一时间范围内升高至32.65%(0.33 mg DOX/mg UiO-66)。当培养基pH值进一步降低至5.0(模拟pH 值范围为4.5~6.5 的内吞区室),释放的DOX 量在2 周后增加到约37.06%(0.37 mg DOX/mg UiO-66)。

图2 UiO-66、UiO-66/Py-PGA-PEG-F3和加载DOX的UiO-66/Py-PGA-PEG-F3的氮吸附-脱附等温线
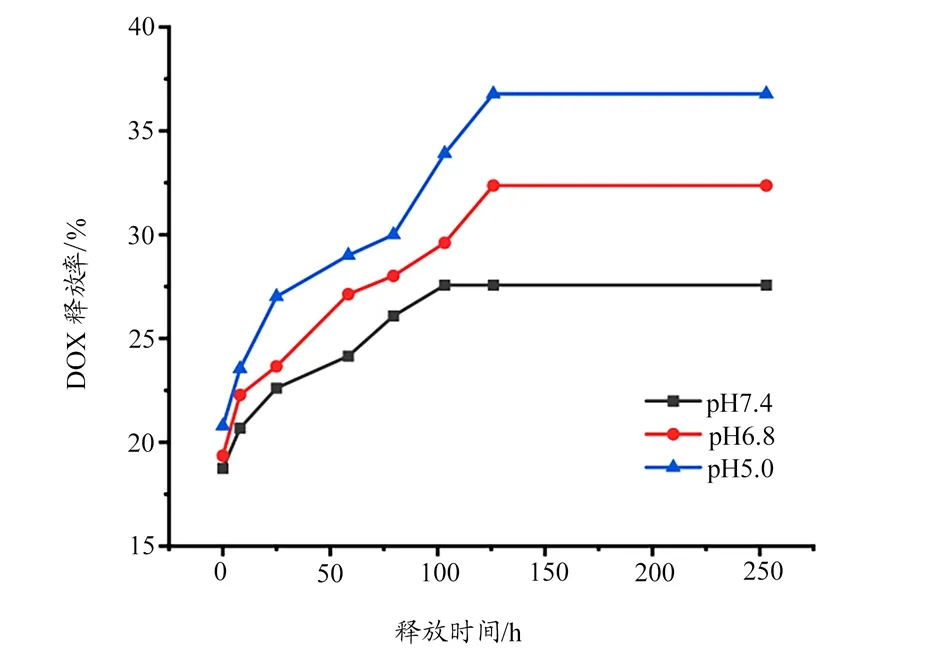
图3 UiO-66/Py-PGA-PEG-F3在不同pH值的DOX释放曲线
DOX@UiO-66/Py-PGA-PEG-F3 的DOX 释放曲线在模拟生理条件下进行测试,不同pH 值对UiO-66 偶联物中DOX 的释放速率有影响[23],并在一定程度上定量分析,结果证实DOX@UiO-66/Py-PGA-PEG-F3 的DOX 释放是持续的并且依赖于pH 值,这与已有的研究结果一致[11]。
2 结果
2.1 体外肿瘤细胞靶向
2.1.1 MDA-MB-231三阴性乳腺癌细胞和L929成纤维细胞特异性结合对比
在肿瘤靶向对比试验中,为验证nMOF 对肿瘤细胞的特异性结合性能,选择MDA-MB-231 三阴性乳腺癌细胞(核仁素+)和L929 成纤维细胞(核仁素-)两个细胞系,用于UiO-66 偶联物的体外评估,通过蛋白质印迹确认核仁素在这两种细胞中的表达谱。主要通过加载在UiO-66/Py-PGA-PEG-F3 或UiO-66/Py-PGA-PEG 上的DOX 作为荧光团指示剂,来显示这些纳米缀合物的位置。流式细胞术和共聚焦显微镜检查的结果显示,在MDA-MB-231 细胞中,DOX@UiO-66/Py-PGA-PEG-F3的荧光强度明显强于DOX@UiO-66/Py-PGA-PEG,同时两种UiO-66 结合物的荧光主要分布在细胞质中。相反,如图4 所示,DOX@UiO-66/Py-PGA-PEG-F3 和DOX@UiO-66/Py-PGA-PEG 与L929 成纤维细胞的非特异性结合程度较低。

图4 MDA-MB-231三阴性乳腺癌细胞(核仁素+)(a)和L929成纤维细胞(核仁素-)(b)与DOX@UiO-66/Py-PGAPEG-F3和DOX@UiO-66/Py-PGA-PEG孵育的代表性共聚焦荧光显微镜图像(均含有50 μg/mL的DOX)
2.1.2 DOX@UiO-66/Py-PGA-PEG-F3和DOX@UiO-66/Py-PGA-PEG对MDA-MB-231细胞的抑制作用对比
为了追踪UiO-66/Py-PGA-PEG-F3 在被MDA-MB-231 细胞内化后的命运,细胞溶酶体被溶酶体(Thermo-Fisher)染色并在显微镜下检查。由于DOX 和LysoTracker 的荧光光谱在荧光显微镜下不可分离,因此使用异硫氰酸荧光素(Fluorescein Isothiocyanate,FITC)代替DOX作为荧光货物。如图5 所示,LysoTracker 的分布模式与FITC 的分布模式高度重合,表明大部分UiO-66/Py-PGA-PEG-F3 在细胞内化后进入溶酶体。在测试浓度范围(0~50 μg/mL)内,UiO-66/Py-PGA-PEG-F3 对MDA-MB-231 细胞无明显的细胞毒性,而DOX@UiO-66/Py-PGA-PEG 和DOX@UiO-66/Py-PGA-PEG-F3 通过释放DOX 抑制MDA-MB-231 细胞的生长。其中,DOX@UiO-66/Py-PGA-PEG-F3 对MDA-MB-231 细胞的抑制作用强于DOX@UiO-66/Py-PGA-PEG。

图5 MDA-MB-231细胞中FITC@UiO-66/Py-PGA-PEG-F3与溶酶体的共定位(用LysoTracker测量)
为了进一步阐明MDA-MB-231 细胞与UiO-66 结合物之间的动态相互作用,将89Zr-UiO-66/Py-PGAPEG-F3 与MDA-MB-231 细胞共孵育。89Zr-UiO-66/Py-PGA-PEG-F3 的细胞内化在0.25 h 内趋于稳定,并且几乎等量的89Zr-UiO-66/Py-PGA-PEG-F3 无须内化即可与MDA-MB-231 细胞表面结合。同时,一旦内化到细胞中,小于30%的89Zr-UiO-66/Py-PGA-PEG-F3 可以在24 h的时间范围内逃逸。由此再次证实了UiO-66/Py-PGAPEG-F3 和MDA-MB-231 之间的特异性结合(可能通过细胞表面的核仁素),为其在体内进一步的研究和探讨提供了基础。
2.2 89Zr-UiO-66/Py-PGA-PEG-F3在MDA-MB-231肿瘤靶向中PET成像对比
利用89Zr 的长半衰期,可以在注射后120 小时内全面检查89Zr-UiO-66 偶联物的分布和衰减曲线。89Zr 是一种具有良好临床潜力的同位素,适用于长期监测不同的代谢过程。从PET 结果的感兴趣区域分析中获得的定量器官分布如图6 所示,89Zr-UiO-66/Py-PGA-PEG-F3 在MDAMB-231 肿瘤中表现出快速积累,这在感染后0.5 h 清晰可见并在2 h 后保持稳定[24]。相比之下,发现未附着F3 肽的89Zr-UiO-66/Py-PGA-PEG 的肿瘤摄取始终显著低于89Zr-UiO-66/Py-PGA-PEG-F3检查点。在晚期时间点(>48 h p.i.),MDA-MB-231 肿瘤中89Zr-UiO-66/Py-PGA-PEG-F3 的摄取随时间逐渐减少。连续PET 扫描,包含MDA-MB-231 肿瘤中心的冠状载玻片,连续采血方法确定89Zr-UiO-66/Py-PGA-PEG 的循环半衰期约为118.8 min(图7)。

图6 89Zr-UiO-66/Py-PGA-PEG-F3、89Zr-UiO-66/Py-PGAPEG-PEG和89Zr-UiO-66/Py-PGA-PEG-F3+阻断剂量F3肽(10 mg/kg小鼠体重)注射后MDA-MB-231荷瘤小鼠不同时间点的典型冠状位PET图像

图7 89Zr-UiO-66/Py-PGA-PEG在裸鼠体内循环半衰期的测定
随着时间的推移,MDA-MB-231 肿瘤中89Zr-UiO-66/Py-PGA-PEG-F3 的摄取减少的原因主要包含两个方面:①89Zr-UiO-66/Py-PGA-PEG-F3 的尺寸较大,可以阻止一些89Zr-UiO-66/Py-PGA-PEG-F3 有效内化到肿瘤(脉管系统)细胞中,导致其从MDA-MB-231 肿瘤中“清除”。因此,更小和更均匀的UiO-66 nMOF 将更有利于临床使用。②89Zr-UiO-66/Py-PGA-PEG-F3 的流出率有助于减少肿瘤积聚。需要阐明89Zr-UiO-66/Py-PGA-PEG-F3 与负责药物或者货物去除的蛋白之间的相互作用,以减少肿瘤中UiO-66 偶联物的损失[25]。随着时间的推移,肿瘤摄取的减少并非由89Zr-UiO -66 nMOF的降解引起的,在5 d(与PET 成像时间相同)内,其在不同的pH 值(低至5.0)下相对稳定。
从PET 图像来看,肝脏和脾脏是捕获89Zr-UiO-66结合物最显著的器官,他们的摄取在3 组数据中都相似(图6)。89Zr-UiO-66 偶联物从肝脏中清除缓慢,肝脏摄取随时间逐渐减少,从感染注射后0.5 h 约50% ID/g 到感染注射后120 h 约40% ID/g,在整个成像时间范围内,未检测到放射性物质沉积在骨骼或肾脏。这一结果再次证实了89Zr-UiO-66 偶联物在体内的优异稳定性,因为89Zr4+离子对磷酸盐具有高度亲和力,易于掺入骨骼中的羟基磷灰石结构中[26]。除了肿瘤摄取的差异外,F3肽的缀合或F3 肽阻断剂量的给药并未改变这些UiO-66缀合物在荷瘤小鼠中的体内动力学,表明核仁素靶向是89Zr-UiO-66/Py-PGA-PEG-F3 增强肿瘤摄取的关键促成因素之一。
3 讨论
与其他纳米载体相比,nMOF 作为生物医学成像和药物递送剂的开发尚处于起步阶段。尽管如此,得益于nMOF 独特的结构和化学多样性、合成条件温和特性以及能够以前所未有的高负载量携带多种成像和治疗剂的能力,其成为一种具有巨大潜力的全新纳米载体平台。
F3 肽在本研究中被选为肿瘤靶向配体,因为其在肿瘤脉管系统中表现出与肿瘤细胞和活化内皮细胞的有效结合,根据Porkka 等[19]的研究,荧光素标记的F3 肽与MDA-MB-435 乳腺癌细胞结合并被其内化,最初出现在这些细胞的细胞质中,然后出现在细胞核中。另外Christian 等[20]的研究表明,F3 肽选择性地与肿瘤细胞表面的核仁素结合,随后可以转运到细胞核和细胞质中。细胞表面核仁素表达升高是各种癌细胞,尤其是乳腺癌的重要特征。将半胱氨酸残基掺入F3 肽的C 末端以促进其与UiO-66 nMOF 的缀合(通过硫醇马来酰亚胺与芘-PEG-马来酰亚胺反应)[21]。
本研究表明,当暴露于模拟肿瘤细胞外pH 值为6.8 时,DOX 的释放百分比在同一时间范围内升高至32.65%(0.33 mg DOX/mg UiO-66)。当培养基pH 值进一步降低至5.0(模拟pH 值范围为4.5~6.5 的内吞区室),释放的DOX 量在2 周后增加到约37.06%(0.37 mg DOX/mg UiO-66)。由此证实DOX@UiO-66/Py-PGA-PEG-F3 的DOX 释放是持续的并且依赖于pH 值,这与Zhao 等[11]的报道一致。
18F-FDG 和64Cu 作为PET 成像显像剂已有较为广泛的研究[27-29]。借助nMOF 的高度可调性,未来将有更多显像剂和药物纳入nMOF 应用范畴。特别是,将成像剂和治疗剂结合至同一nMOF 平台,有望大幅度促进这类具有发展前景的治疗诊断纳米颗粒的功效研究。nMOF的研究与应用亟待进一步优化,通过增强其生物相容性成分和表面功能,从而确保其在延长血液循环、逃避网状内皮系统及组织特异性方面的表现。尽管已对nMOF进行了大量的体外药效研究,但体内药效的系统研究尚未展开。为了优化nMOF 的性能,有必要开展相关体内研究,经过优化的nMOF 在生物医学成像和药物输送领域具有广阔的应用前景。
4 结论
本征放射性UiO-66 nMOF 材料(89Zr-UiO-66)被设计、合成和表面工程用于肿瘤靶向和增强药物递送,本研究验证了其良好的放射化学稳定性、材料完整性和可靠的材料功能化,可以将相对大量的DOX(1 mg DOX/mg UiO-66)加载到UiO-66 偶联物上,但DOX 释放行为需要进一步优化。通过采用F3 肽(靶向核仁素)作为靶向配体,89Zr-UiO-66 结合物对MDA-MB-231 肿瘤的特异性和显著增强靶向性在体内得到证实,这在离体实验中得到了进一步证实。中剂量和大剂量UiO-66/Py-PGA-PEG-F3 的毒性研究未显示出显著的体内毒性。此外,在荷瘤小鼠中也证实了增强的DOX 向体内MDAMB-231 肿瘤的递送。当前较多的是尝试通过改进表面工程方法和研究纳米缀合物用于联合放化疗的潜力来进一步优化它们的体内药代动力学,希望其他研究人员将不同类型的放射性nMOF 材料用于癌症治疗应用。
