遗传性血色病的研究进展
2024-05-08韩悦张欣欣
韩悦 张欣欣
遗传性血色病(HH)是一种异质性很强的遗传性疾病,表现为组织中铁沉积过多。排除后天原因,如输血、各种导致铁过载的贫血、慢性肝病、肿瘤、感染、系统性红斑狼疮、类风湿关节炎所致的继发性铁沉积后,均应考虑HH[1]。
铁稳态对于人体至关重要。食物中的铁在十二指肠上皮细胞被吸收入血,过程涉及一系列蛋白:细胞色素B(cytochrome B)、二价金属转运体1(divalent metal transporter 1)、运铁素(ferroportin)和亚铁氧化酶[或称膜铁转运辅助蛋白(hephaestin)]。随后通过转铁蛋白(transferrin)经血循环运输到包括肝细胞、网状内皮系统(RES)等拥有转铁蛋白受体的靶细胞,细胞借内吞作用摄入铁,以铁蛋白(ferritin)形式沉积在组织中[2]。
铁调素(Hepcidin)是由肝细胞产生、调节循环铁含量最重要的激素。其与运铁素结合后致运铁素降解,减少铁从肠道吸收,从肝细胞、RES释放到循环中,以维持铁稳态。影响铁调素能导致铁的吸收失调和组织中铁蓄积,而HH致病机理大多涉及铁调素缺陷。
铁沉积导致的器官损伤常见于肝脏、心脏、内分泌系统、关节和胰腺。西方一般按照是否为HFE基因致病,分为HFE型和非HFE型。过去将非HFE型分为3型,涉及4种致病基因[3],最新的欧洲血色病指南非HFE型基因名单中加入了CP、TF、BMP6基因[4]。
由于中西方致病基因差别较大,我们曾在2019年对HH的基因诊断结合我国数据进行了系统性的综述[5]。近年来国际各大指南先后出现了少见的更新,且国内临床上对HH的重视度明显增高,新的研究数据也在稳步产出。本文希望通过对已有知识进行一个更新,为处理不明原因铁过载提供一些新线索。
一、HH致病基因
1.HH1型(HFE-HH)
该型为经典HH,是由HFE(H代表高,FE代表铁)基因变异引起的常染色体隐性遗传。西北欧凯尔特语系人种中该基因C282Y变异最多见,青铜时代开始出现在英国样本中[6]。该变异的纯合型铁过载的风险最高,其次为H63D变异,其他罕见突变报道多见于拉丁语系国家,本文中不再赘述。发病率最高的欧洲国家包括爱尔兰、英国、挪威、法国、葡萄牙和丹麦[7]。欧洲、美国、加拿大HH指南描述的一般均为此型,其中有专家建议普查C282Y[1,3-4,8],但至今仍未实施。HFE基因C282Y的外显率可能会受到其他基因影响。UK Biobank 2022年的一项外显率研究发现在2 890例患者中,铁代谢相关多基因突变可显著增加临床外显率,潜在修饰基因包括BMP2、BMP6(见下文)、GNPAT、PCSK7等,提示修饰的复杂性[9]。该型表现多样,90%的患者无症状。男性外显率更高、发病早,可能与女性在月经期间铁流失有关。
若有下述脏器累及,导致出现如疲劳、关节疼痛、转氨酶水平升高、肝脏肿大、不明原因的心肌病时应怀疑。内分泌异常包括糖尿病或性腺功能减退,原因分别是铁沉积在胰腺和垂体[1]。约15%的患者心脏受累,但只有约3%会发展成心肌病,同时伴心律失常和心力衰竭的风险[1,10]。目前尚无评估患者患有心肌病的铁蛋白阈值,故临床诊疗应评估患者的心脏症状,如胸痛、下肢肿胀和气短,同时可考虑进行心电图和超声心动图检查,以筛查心律失常;对于检查结果呈阳性的患者,可通过MRI诊断心肌铁沉积[4]。约25%的患者肝脏受累,血清铁蛋白水平超过1 000 ng/ml的患者发生晚期肝纤维化的风险最高,未经治疗的男性患者一生中患肝硬化/肝细胞癌(HCC)的风险约为9%[1,10]。70%晚期患者会出现皮肤色素沉着[1]。骨关节表现常以非特异性关节疼为首发症状[11],通常累及第2或第3掌指关节[12]。累及中枢神经系统方面:2022年的两项研究显示C282Y纯合子患者的脑部,尤其是皮层下运动结构的铁含量增加[13]。在UK biobank中关于中老年人数据显示,C282Y纯合子与肌肉疲软、虚弱和慢性疼痛有关;同一数据库中对2 889例C282Y纯合子和485 399例对照者比较,发现男性携带者的运动障碍患病率显著增加[14]。但在31例运动障碍的C282Y男性同卵双生者中,仅10例同时被诊断为HH[13]。
除此之外,近年来合并迟发型皮肤卟啉症(PCT)的患者也进入了临床视野。近期由英美研究团队报道的1例女性患者四肢出现光敏性皮疹和水疱,使用类固醇类药物后症状未见好转,伴多发性关节痛和肝功能异常,后确诊PCT合并HH1,定期放血治疗后病情有所好转[15]。二者之间是否有因果关系,尚不明确。
国内HH主要为非HFE基因所致,2021年有研究对32例铁过载患者的基因筛查发现97%均为非HFE致病基因,以HJV和SLC40A1多见[16]。我中心研究团队收集的我国人群数据显示,该基因变异在铁过载人群中出现概率极低,符合既往中外报道。
2.HH2型
该型起病较早、发病率低,多见于近亲后代,可分为2型,由不同基因突变导致:
(1)2A型:又名HFE2,由编码hemojuvelin的HJV基因突变所致,常染色体隐性遗传,出现铁积累较早,导致发病年龄提前,常伴心肌病、糖耐量降低和性腺功能减退等内分泌功能衰竭,肝脏受累不严重。幼年型病例常见于意大利[17]、希腊和法裔血统加拿大人群。
近年有出现中年亚洲患者的报道[5]。2022年广州学者报道了1例31岁女性,铁沉积发生在患者的心脏、肝脏、内分泌器官及皮肤,相应引起心律失常、转氨酶升高、糖尿病、闭经、骨质疏松症及色素沉着,HJV基因变化为C89Y纯合突变,家系筛查证实突变分别来自父母[18]。同样,在我中心出现的2例患者也均为中年发病,但均为杂合突变,且伴发铁代谢通路其他基因缺陷,无法确认致病性。
(2)2B型:由编码铁调素的HAMP基因突变所致,常染色体隐性遗传,致病突变直接导致铁调素缺陷,国内尚无相关数据。考虑到该基因的重要性,一旦出现问题,则直接影响胚胎发育,故出现成年患者几率微小。因此面对低龄患者及症状重时可考虑为该型。
3.HH3型
由编码转铁蛋白受体2的TFR2基因突变导致,罕见发病,常染色体隐性遗传,报道多见于拉丁族裔,也有日本病例报道,症状介于HH1型和HH2型之间,包括肝功能异常、肝纤维化、肝硬化、糖尿病、性腺功能减退、心肌病和皮肤色素沉着。2022年报道了1例我国HH3型患者,症状累及肝脏和皮肤,检测到复合杂合突变G430R/Y320X,在家系中先证者和其胞姐具有相同突变,但表型不同[19]。我中心虽测得TFR2罕见突变,但均为杂合突变,且位于非编码区。
4.HH4型
由编码运铁素的SLC40A1基因缺陷导致,该基因负责将二价铁从肠道、肝脏、RES的细胞内转运到细胞外,控制饮食中铁的吸收、巨噬细胞和红细胞对铁的回收及肝细胞中储存铁的释放。当血清中铁过量时,循环中的铁调素水平会升高,导致SLC40A1降解,从而限制铁向血液外流出[20]。按照对运铁素功能的影响可分为两型,遗传方式目前均认为属于HH中少见的常染色体显性遗传:
(1)4A型-巨噬细胞型[又称运铁素病(FD)]:由SLC40A1基因功能缺失突变导致,是西方非HFE-HH最常见原因之一。SLC40A1基因功能缺失影响运铁素输出功能,阻止铁从RES中释放,导致脾脏铁沉积增加,肝脏铁沉积适度增加[21],影像学可见“黑脾”。体内铁储存量并未大幅增加,临床表现可能较轻,血清转铁蛋白饱和度水平(TS)通常正常。国内团队报道的T419Ⅰ杂合突变出现在49岁男性患者,高铁蛋白、TS正常、脾脏及肝脏铁沉积不明显,合并纯红细胞增生症和淋巴细胞白血病,铁螯合剂治疗有效[22],不过同样携带该突变的患者女儿尚未出现铁代谢异常,且报道中未显示家族中有其他阳性病例,因此我国人群的遗传方式有待考证。该型一般不建议放血治疗。
(2)4B型-肝型:由SLC40A1基因的功能获得突变导致,临床表型近似经典HFE-HH。北京研究小组分析22例铁过载患者后发现3例存在Y333H,其中2例可明确家族史,体外实验证实变异致运铁素对铁调素产生抗性[23]。天津团队研究报告了1例Y333H杂合患者接受了为期一年频率为每月1次的红细胞分离治疗,其后口服铁螯合剂后好转,该病例有明确家族史,基因检测确认遗传方式[24]。杭州团队报道了2例老年兄弟患者,均有肝硬化、糖尿病、皮肤色素沉着、高铁蛋白血症和高TS,影像学检查结果显示肝脏及脾脏均有铁沉积,铁螯合剂治疗有效,兄弟二人均检出C326Y[25],该突变2004年在东南亚患者中被发现[26],国内最早在2015年由广州团队报道[27]。日本团队发现的1例H507R杂合突变,铁调素水平、铁蛋白、TS增高,肝脏铁沉积较明显,但未见“黑脾”描述,且放血治疗有效[28]。此型目前在国内报道中较多见。我中心出现的2例患者均为青少年发病,但无明确家族史及贫血。
除我国人群的遗传方式待定外,临床上并非所有SLC40A1基因缺陷都能明确分型,如吉林团队报道了1例34岁的V162del缺失突变男性患者,首发症状是肝区不适、铁蛋白高、肝脏及脾脏铁沉积,但TS正常且放血治疗有效[29]。
5.新版国际指南增加的推荐基因
新收录建议检测的基因包括CP、TF和BMP6[4]:
(1)CP基因:编码铜蓝蛋白,可导致铜蓝蛋白缺乏或功能障碍,罕见发病,常染色体隐性遗传。铜蓝蛋白是一种铁氧化酶,参与氧化亚铁,随后铁以这种形式被转铁蛋白转运。铜蓝蛋白缺乏导致铁积聚在胰腺、肝脏和神经组织中。该病特点是会影响神经组织。患者表现为小细胞低色素性贫血、铁蛋白升高[13]、铜蓝蛋白量极少甚至没有。明确出现无或低铜蓝蛋白血症时需进行基因检测。其致病机理尚未厘清,理论上铁分布应该类似FD[21],TS通常正常。意大利一项非酒精性脂肪肝队列研究中,铜蓝蛋白缺陷与高铁蛋白水平、肝脏铁沉积和严重肝病有关[30]。国内未见报道,上海本地数据中也没有发现CP基因突变,但确实出现过HH致病基因合并肝豆状核变性致病基因突变的情况,提示可能存在两种金属代谢通路缺陷之间的联系,鉴于该类铜代谢异常遗传疾病在国内发病率不低,怀疑HH且常规基因无法解释时可考虑检测。
(2)TF基因:编码转铁蛋白,导致外周血转铁蛋白下降,常染色体隐性遗传。红细胞依赖其结合铁,该基因缺陷导致血浆转铁蛋白水平极低,小细胞低色素性贫血,影响发育,完全缺失导致高致死率,因此临床上罕见。非转铁蛋白结合铁是此类患者血浆铁的主要形式。此外,由于贫血,血钙素的表达也会降低,从而导致铁的吸收率提高,这与血浆中的非转铁蛋白结合铁一起,导致肝脏、胰腺和心脏的铁负荷过重。TS高则血清铁高。补铁、输血无效,反而会加重病情。有条件的患者应补充去铁转铁蛋白配合螯合剂使用。国内近10年无报道。我中心研究小组出现过1例非编码区罕见突变,铁过载并出现低转铁蛋白,同时还存在铜代谢基因异常,但由于杂合不支持隐性遗传,其致病性未能确定。
(3)BMP6基因:编码骨形态发生蛋白6,属于转化生长因子-β强效多功能生长因子家族,结构高度保守。肝脏铁过载可诱导BMP6基因表达,并激活BMP/SMAD通路[31]。影响BMP6前肽的杂合子突变与铁调素水平异常降低、某些人群的轻度铁过载有关[32]。国内暂无报道。我中心研究小组10年来仅有1例杂合编码区突变铁过载患者,同时还存在其他铁代谢基因异常。遗传方式未明确。
6.新增候选基因
2022年德国研究者发现3例男性患者的异常表型与幼年HH型相似,并伴有严重的神经系统异常(早发癫痫、严重发育迟缓和智力障碍),在童年时期因神经系统检查结果而被确诊,铁负荷过重、血红素水平低,均存在PIGA基因变异[33]。北京团队发现的一系列新的非HFE型致病基因,如UBE2O、PCSK7[34]、DENND3[35]基因。此外,还有NMBR基因[36]。其他候选基因包括红细胞相关致病基因的个案报道,严格意义上不属于HH范畴[37-38]。上述基因致病性尚待验证。
二、特殊临床表型
1.眼部症状:遗传性高铁蛋白血症-白内障综合征(HHCS)是一种罕见的、经常被误诊的常染色体显性遗传病,以血清铁蛋白水平高和幼年双侧白内障为特征,通常不伴有铁过载。由FTL(铁蛋白L亚基)基因的铁反应元件突变引起[39]。该区域突变会影响铁调控蛋白(IRP)与该区域的结合力,使L-铁蛋白的表达失调,合成增加。至2023年,全球白内障版图数据库(Cat-Map)报道FTL基因缺陷致病的家系不过120余例[40]。近期一项巴西研究对来自3个不同家族的8例HHCS及家系中1例临床症状阴性成员进行了分析,在所有罹患眼疾的个体中均发现了FTL的可能致病变异c.-157G>A[41],其均在14岁前出现缓慢进展的双侧白内障症状,表型为不同的双侧弥漫性白内障,且均出现高铁蛋白血症;有两例患者同时检测出HFE致病突变(H63D),铁蛋白值也高于其他患者。同时存在FTL和HFE基因的致病突变是否会导致更高的高铁蛋白血症,还需要更多的病例支持。
对于不明原因高铁蛋白血症和双侧白内障患者或早期视力减退,当血清铁浓度和TS正常时,需考虑此型。目前在我国尚未见报道,但我中心曾检测到5例H亚基罕见突变,其中1例为高加索人且致病性未明。
2.感染性疾病:由于铁蛋白升高是感染急性期反应的一部分,和HH一样,感染性疾病也需获得其数值用于评估疾病进展。在COVID-19大流行期间,也出现了意外发现无症状HH的报道,特点是在中度感染期间铁蛋白水平极度升高[42]。其他病原体感染,如对于隐球菌病来说,肝硬化、糖尿病和铁过载本来就是该病的独立风险因素,而HH反过来又可直接导致上述3种情况。近年来确有报道提示隐球菌合并HH的情况[43]。虽然目前支持COVID-19或其他感染和HH的因果关系的证据较少,但提醒我们感染可导致铁蛋白明显升高,而极度升高不一定仅是感染所致。
三、临床评估及新技术
1.铁蛋白和TS
约98%的HH1患者的TS水平≥45%(正常参考范围20%~45%)。不过年轻患者和非HH1型患者的TS水平可能<45%。铁蛋白水平在炎症状态下,包括其他肝脏疾病、饮酒和肥胖均可能增高,从而导致假阳性[1]。
2.肝脏组织病理活检(简称肝活检)
肝活检曾是诊断金标准,随着对疾病认识的提高,诊断越来越多地基于基因检测、新型成像技术判断[2]。2022年新版欧洲肝病研究会指南建议,当铁蛋白>1 000 ng/ml时,可选择肝活检来评估肝硬化或肝纤维化,除非肝硬化的临床表现非常明显,或可通过瞬态弹性成像技术无创地显示肝纤维化[4]。
3.MRI
近年出现的非造影剂增强MRI与估算肝铁浓度的软件相结合可无创评估铁沉积[44]。目前已开发并验证了3种主要技术:信号强度比、R2和R2*法。但存在如图像噪声和肝内脂肪等干扰因素。混杂因素校正的R2*的肝铁浓度是目前最实用的方法[45]。不过该类方法由于缺乏准确评估放血治疗的时机,降低了其临床实用性。肝铁浓度测量方法仍在继续开发优化中,这些技术需要进一步的验证和监管机构的批准才能广泛使用[46]。
4.基因检测
仅当临床怀疑HH时,才需要进行基因筛查。如上文所述,最新欧洲指南建议评估HH的最小基因集至少应包括HFE、HAMP、HJV、TFR2、TF、CP、BMP6、SCL40A1等基因[4]。如有条件,临床全外显子组测序或靶基因测序可用于涵盖更多的候选基因,但结果的解读可能具有挑战性[47]。一旦基因检测结果为阳性,可有助于对疾病进行分期:基因阳性,但无铁过载为1期;基因阳性且有铁过载,但无器官损伤为2期;基因阳性且有铁过载和器官损伤为3期。分型有利于临床医生监测疾病的进展情况[3]。
四、治疗
放血治疗仍是一线疗法,适用于TS水平>45%,且铁蛋白>300 ng/ml(男)/200ng/ml(女)。有症状和铁蛋白水平>1 000 ng/ml的患者获益最大。治疗的目标是使患者铁蛋白水平达到50~100 ng/ml且不出现贫血,则每周放血需要持续数年。达到目标值后,频率可减少到每年3~4次。对于1期患者,可每年评估其TS和铁蛋白水平[4]。放血治疗降低了肝硬化和HCC的风险,改善了肝功能、心功能障碍和疲劳。在106例接受治疗的晚期纤维化患者中,38.6%的患者在中位9.5年的治疗随访时间内纤维化程度降至早期,而早期及晚期纤维化患者的HCC发病率分别为2.3‰和32.8‰。放血治疗不能改善糖尿病、性腺功能减退症或关节痛。肝衰竭或HCC患者应进行肝移植,这是唯一的治愈方法,可永久性地使铁吸收正常化。不过上述数据均基于HH1型患者产生[48],我国的非HFE-HH相关放血治疗数据仍缺乏。
口服铁螯合剂在HH中证据不足,仅对放血治疗不耐受、无反应、有潜在危害的患者(如严重贫血、心力衰竭),在权衡不良反应和收益后考虑使用。质子泵抑制剂会增加胃的pH值,从而减少铁的吸收,如有其他病症,可使用质子泵抑制剂作为辅助治疗[4]。
五、总结
HH是一种异质性很强的引起铁过载的遗传性疾病。东西方人群致病基因谱完全不同。可结合临床症状、基因检测、生化及影像学检查结果来指导诊断思路(图1)。治疗上,虽然放血治疗可大大降低发病率和死亡率,但不适用于个别亚型,甚至会起到反作用。建议在临床诊断困难的时候,对患者和直系亲属进行全基因或全外显子组测序,并结合有HH领域经验的中心人员进行解读。
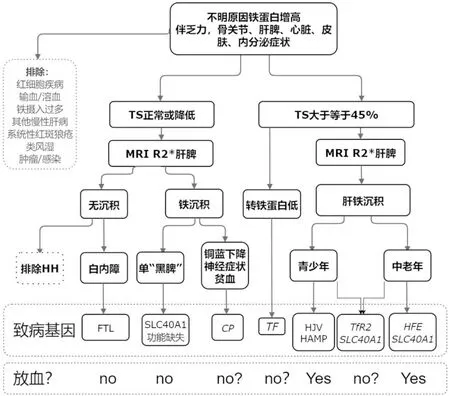
图1 适用于国内患者的临床诊断思路导向图
