红景天苷改善创伤性脑损伤后继发性神经损伤的作用机制研究进展
2024-05-06葛红飞喻安永
陈 琳, 葛红飞, 喻安永
(1.遵义医科大学附属医院 急诊科,贵州 遵义 563003;2.陆军军医大学第一附属医院 神经外科,重庆 400037)
创伤性脑损伤(traumatic brain injury, TBI)是由外部机械力对颅脑直接撞击造成的开放性或闭合性损伤,是导致青年人群死亡和残疾的最常见原因之一,尤其在第三世界国家及发展中国家中多见[1]。据统计,中国基于人群的脑外伤死亡率约为13/10万,与其他国家报告的死亡率相似[2]。TBI因高致死率和致残率,已经成为严重影响全球人民生命健康的公共卫生问题。TBI后局部脑组织水肿和血肿引起颅内压升高,脑血流相对下降,导致神经细胞缺血缺氧或直接死亡。而坏死的脑组织和炎症介质释放,加剧局部炎症反应、氧化应激(oxidative stress, OS)、神经元凋亡和轴突变性,进而导致神经功能障碍的病理生理并发症[3-5]。其中继发性脑损伤(secondary traumatic brain injury, sTBI)导致的神经功能障碍一直是临床上治疗TBI的重点和难点,亦是导致患者高致残率的最主要原因。
红景天(rhodiola rosea)是广泛分布于欧洲、亚洲和美国的一种天科类植物,红景天苷(salidroside, Sal)是其主要生物活性成分之一,在我国传统医学中被广泛用于预防和治疗各种疾病。在药代动力学中,Sal显示出快速吸收和消除,在非生理条件下Sal的生物利用度升高,该成分及其代谢物(酪醇)能够分布到脑组织并保持较高的浓度[6]。而且,在各种动物实验和临床试验中Sal几乎没有明显的毒性或副作用[7-8]。已有研究表明,正常大鼠在静脉或口服给药后,Sal在大脑中呈低水平分布[9],TBI后放大的促炎机制以及内皮紧密连接蛋白表达下降[10],导致血脑屏障(blood brain barrier, BBB)开放[11-12],大量Sal进入脑组织后,通过抑制NF-κB/NLRP3通路下调脑组织中炎症小体释放,促进脑组织中能量代谢和BBB中紧密连接蛋白闭塞带-1(zonula occludens-1, ZO-1)、闭塞蛋白(occludin)和claudin-5的表达[13],达到改善神经传导功能、减轻神经炎症,促进神经再生和屏障功能恢复的作用[14-17]。虽然针对TBI各国都采取了相应措施(如配戴头盔、安全带立法等),但TBI在全球的发生率仍呈上升趋势[18]。而Sal作为一种对中枢神经系统有益的治疗药物,如何恢复TBI后神经活性及功能,其作用机制还尚无总结,因此本文就其在sTBI中促进神经功能恢复的作用机制进行综述,以期为sTBI的临床治疗提供借鉴和参考。
1 Sal通过抑制氧化应激反应促进sTBI神经功能恢复
sTBI是在原发颅脑损伤的基础上,一系列生化级联反应诱导并加重的脑组织损伤。它产生的OS是伴随TBI整个过程并导致认知功能障碍的最重要原因之一。sTBI患者体内H2O2和活性氧自由基(reactive oxide species, ROS)含量持续升高,抗氧化酶产生减少[19-20],进而诱导脂质过氧化、核和线粒体DNA损伤、神经元凋亡、BBB破坏和炎症介质大量释放[21-22],导致脑组织中小胶质细胞广泛激活。过量的ROS和活化的小胶质细胞通过激活非经典核因子-κB(noncanonical nuclear factor-κB, NF-κB)和分泌烟酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide phosphate, NADPH)氧化酶[23],促进白细胞介素1β(interleukin 1β, IL-1β)、白细胞介素6(interleukin 6, IL-6)和肿瘤坏死因子-α(tumor necrosis factor-alpha, TNF-α)、ROS等过量产生[24-25],形成炎症-氧化级联反应。此外,ROS与神经元一氧化氮合酶(neuronal nitric oxide synthase, nNOS)以及突触后密度蛋白95(postsynaptic density protein 95, PSD95)相互作用,形成高活性的过氧亚硝酸盐,其与突触表面谷氨酸能相关蛋白n-甲基-d-天冬氨酸(n-methyl-d-aspartate, NMDA)受体的酪氨酸残基相互作用,导致神经元细胞功能障碍[1]。随后,小胶质细胞介导的神经炎症和OS产生慢性和进行性神经退行性改变,导致晚期神经功能障碍和认知缺陷[26-27]。由此可见,sTBI源于ROS、炎症介质、过氧化物的过量产生,其中ROS与nNOS以及NMDA受体的物理偶联是氧化损伤的关键。这种关联表明,sTBI的抗氧化系统功能障碍可能与损伤后突触病有关。
Sal是一种生物活性提取物,主要来自传统药用植物红景天,其凭借更高的疗效和安全性,已成为预防和治疗sTBI的潜在神经保护剂。已有研究发现,Sal能显著减小脑梗塞面积,预防脑水肿并改善神经功能,对缺血性卒中具有强大的治疗作用[7]。sTBI由于局部脑组织缺血缺氧、坏死,大量炎症因子释放,导致机体产生大量的ROS,而过度富集的ROS诱导线粒体嵴扩张、线粒体外膜破裂和细胞色素C释放[28],受损线粒体进一步释放到细胞质中,激活半胱天冬酶依赖性凋亡级联事件,最终导致神经元死亡。值得我们关注的是,已有研究证明Sal诱导大脑中动脉闭塞(middle cerebral artery occlusion, MCAO)大鼠脑组织中超氧化物歧化酶(superoxide dismutase, SOD)、谷胱甘肽过氧化物酶(glutathione peroxidase, GSH-Px)和谷胱甘肽-S-转移酶(glutathione-S-transferase, GST)的活性升高[29-31],降低丙二醛(malondialdehyde, MDA)和8-羟基-2′-脱氧鸟苷(8-hydroxy-2′-deoxyguanosine, 8-OHdG)、NADPH氧化酶(NOx)水平,抑制ROS的产生[32],并通过增强PINK1-Parkin信号通路,促进线粒体自噬,消除受损的线粒体[33],进而显著减轻缺血再灌注和神经退行性疾病导致的神经功能障碍。同时研究证明,Sal通过抑制NF-κB途径和内质网应激来抑制脂多糖(lipopolysaccharide, LPS)刺激的BV-2细胞中小胶质细胞由M1型极化为M2型,使BV-2细胞中炎症酶的mRNA和蛋白质表达降低,降低诱导性一氧化氮合酶(inducible nitric oxide synthase, iNOS)、环氧合酶-2(cyclooxygenase-2, COX-2)和前列腺素E2(prostaglandin E2, PGE)的产生,增加脑源性神经营养因子(brain-derived neurotrophic factor, BDNF)、cAMP反应元件结合蛋白(cAMP response element binding protein, CREB)、神经生长因子(nerve growth factor, NGF)等表达[34],解除nNOS与NMDA受体的物理偶联,从而降低神经炎症反应和OS,促进中枢神经系统(central nervous system, CNS)损伤后神经再生[28,35-36]。另外,Sal还可以抑制TNF-α和PI3K/Akt/NF-κB等表达,降低OS、炎症反应和细胞凋亡,从而发挥神经保护作用[37-39]。由此可见,OS是sTBI神经功能障碍的重要原因之一,Sal通过增加抗氧化酶的表达,抑制氧化因子的产生,可有效降低了sTBI的神经损伤和神经退行性病变。
2 Sal通过抑制炎症反应促进sTBI神经功能恢复
众所周知,神经源性神经炎症是中枢神经系统维持代谢平衡,以及许多神经系统疾病(包括TBI)的病理发展和进展的关键组成部分[40-41],对TBI愈后有重要影响。sTBI引起的慢性炎性反应长达数周甚至数年,常引发脑部一系列慢性神经炎症级联反应,导致神经退行性病变和死亡。sTBI死亡神经元的碎片刺激神经胶质细胞内Fis1升高,介导小胶质细胞内线粒体碎裂释放至胞外,并激活为M1型[42],进而激活幼稚星形胶质细胞极化为A1型并吸引小胶质细胞迁移到炎症部位,两种细胞分泌多种炎性细胞因子,包括TNF-α和IL-6,以及细胞粘附分子和蛋白酶等。过量的炎症介质作用于BBB,导致其通透性增加,而大量TNF-α浸润降低突触表面氨甲基磷酸(aminomethyl phosphonic acid, AMPA)受体GluR2亚基的表达,导致细胞内Ca2+超负荷诱导神经元死亡和变性。同时,小胶质细胞补体受体3(complement receptor 3, CR3)激活后,从分支状变形为变形虫形状,促使局部吞噬作用和促炎功能增强[43],而激活的NADPH氧化酶与突触表面AMPA受体结合,触发突触传递的长期抑制[44],导致神经血管、血脑屏障和神经元损伤,最终出现认知缺陷的发生和神经变性[45-47]。此外,Perez等[48]发现TBI后海马神经元下调 d-丝氨酸水平,增强星形胶质细胞d-丝氨酸的强直释放,激活钙调磷酸酶/活化T细胞核因子(nuclear factor of activated T cells, NFAT)信号通路,导致神经元的突触强度降低和突触蛋白损失(PSD95和GluR1),加重神经炎症反应。而利用腺相关病毒(adeno-associated virus, AAV)抑制钙调磷酸酶/ NFAT通路后,可以发现突触前因子SPARCL1(hevin)的表达显著增加,突触功能部分恢复,神经炎症也得到缓解[49]。综上所述,神经炎症反应与小胶质细胞和星形胶质细胞的激活状态密切相关,细胞的极化状态影响了神经突触的恢复,决定了sTBI预后。因此,揭示小胶质细胞和星形胶质细胞的极化机制,在sTBI的治疗中尤为重要。
在sTBI病程发展过程中,炎症反应亦是造成神经功能损伤和细胞死亡的重要原因之一。已有研究发现,sTBI产生的炎症介质通过抑制基质金属蛋白酶(matrix metalloproteinase, MMP)的活化、诱导紧密连接蛋白复合物的变异和转运蛋白下调,促使BBB和血管内皮渗透性升高。Sal激活SH-SY5Y 细胞中 DJ-1 -Nrf2 抗氧化途径抑制MMP-9的活化[50],逆转紧密连接蛋白(如claudin-5和occludin)的减少,从而改善BBB和血管内皮渗透性、增强缺血后海马CA1区域的神经发生和功能重建[51]。另外,Sal诱导间充质干细胞 (mesenchymal stem cells,MSCs) 分化为多巴胺能神经元,并增加 DA 释放[52],从而抑制神经炎症和促进神经功能恢复[53-54]。同时,Sal通过激活PI3K/Akt信号通路,促进Akt磷酸化水平和线粒体Bcl-2/Bax比值的增加[55],在氧-葡萄糖剥夺(CM-OGD)条件下,Sal激活Akt/GSK-3途径,刺激大脑胶质细胞增生[56-57],极化小胶质细胞为M2型,星形胶质细胞为A2型,从而显著抑制炎症因子的释放并促进微管相关蛋白2(microtubule-associated protein 2, MAP-2)的表达[58],进而阻止脑内皮中C3及其活性片段C3a的增加,降低大脑内皮胶质细胞活化和中性粒细胞浸润,在OS后恢复抗炎内皮表型[59]。Xiao等[60]研究发现,Sal激活SIRT1通路增加自噬相关蛋白(Beclin-1和P62)的表达,降低炎症因子(TNF-降、IL-1降和IL-6)和ROS产生,以及凋亡相关基因表达(BAX和BCL-2),显著缓解脓毒性脑病小鼠神经炎症和神经损伤。由此可见,Sal可以促进sTBI神经细胞再生,抑制神经炎症和神经细胞凋亡。虽然部分机制已被揭示,但其如何调节炎症反应的机制尚不完全清楚,仍需要进一步探索。
3 Sal通过抑制凋亡,缓解谷氨酸神经毒性反应诱导的神经功能障碍,促进sTBI神经功能恢复
谷氨酸兴奋性神经毒性反应是sTBI的早期事件,是加速缺血损伤区域细胞死亡的重要原因。谷氨酸是脊椎动物神经系统中最丰富的氨基酸,主要由神经元和星形胶质细胞中的谷氨酰胺通过磷酸盐依赖性线粒体谷氨酰胺酶的作用,以及谷氨酸脱氢酶对α-酮戊二酸的转氨酰胺反应合成,从所有兴奋性突触中释放出来[61]。sTBI的缺血损伤区域细胞裂解,细胞外谷氨酸水平迅速升高,游离谷氨酸作用于正常突触后膜的2个主要神经递质受体:NMDA受体和AMPA受体,导致突触后膜离子门控通道开放,促使Na+内流和Ca2+超载,细胞骨架破坏,线粒体膜电位崩溃,电子传递链异常,导致突触后神经元去极化和神经炎性改变、死亡[1]。另外,缺血损伤后线粒体膜电位的改变导致ATP水平降低,无法满足其较高的能量的需求,促使细胞内Ca2+超载和ROS的应激损伤,加剧一系列促凋亡物质的产生,如一氧化氮(nitric oxide, NO)、超氧化物(O2-)和过氧化氢等,最终导致神经元死亡[28]。由此可见,谷氨酸神经毒性反应是sTBI神经细胞死亡的重要途径,细胞外基质中谷氨酸富集和Ca2+超载引起细胞骨架破坏、线粒体崩解、电子传递链异常和神经细胞死亡。而如何干预这一级联反应,减少sTBI谷氨酸释放和线粒体损伤,及时清除凋亡介质和受损线粒体,维持细胞内环境的稳态,是sTBI治疗面临的最大挑战之一。
尽管sTBI损伤的病理生理学机制多种多样,但大部分学者认为神经元死亡是由细胞凋亡诱导的。正常情况下谷氨酸主要来源是突触的溢出,以及星形胶质细胞和小胶质细胞释放。而星形胶质细胞和神经元上的兴奋性氨基酸转运蛋白(excitatory amino acid transporter, EAAT)通过再摄取去除组织中过多的谷氨酸,从而维持细胞内外谷氨酸平衡。sTBI由于星形胶质细胞变性和死亡,其表达谷氨酸转运蛋白(EAAT1和EAAT2)的比例相对下降,引起线粒体膜断裂和活性降低、氧化还原系统失衡、钙稳态破坏,导致细胞外液(extracellular fluid, ECF)的谷氨酸值立即升高,重吸收障碍,进而引起神经兴奋性毒性反应,甚至进一步扩大病理组织的体积[62]。已有研究发现,Sal激活Nrf2 / HO1、PINK1 / Parkin和Sirt1/p53/Drp1信号通路,抑制过量Ca2+流入[38],增加星形胶质细胞中线粒体自噬通量[63],诱导原发性小胶质细胞极化为M2表型,增强其吞噬作用并抑制炎性细胞因子释放[58,64]。通过上调Glu损伤的HT22细胞的细胞活力、ATP水平、Na+-K+-ATP酶活性和线粒体膜电位(mitochondrial membrane potential,MMP)稳定性,降低脂质过氧化和ROS水平,减少缺氧诱导的线粒体裂变,增加线粒体管状或线状形态,从而维持线粒体动力学稳态,有效地保护神经元免受谷氨酸诱导的兴奋性毒性,起到促进神经细胞再生和恢复神经活性的作用[65-67]。上述研究表明,Sal对Ca2+水平的强调节作用,不仅可以减弱ROS对线粒体的损伤,促进线粒体自噬,维持线粒体稳态;而且可以调节小胶质细胞和星形胶质细胞表面标志物的表达,增强其吞噬和抑炎作用,有助于保护神经元免受缺血性损伤引起的神经兴奋性毒性损伤。相关机制总结见图1。
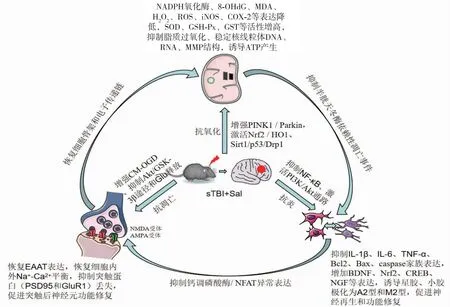
图1 红景天苷改善sTBI神经功能障碍的相关机制
4 展望
本文综述了Sal在sTBI中的抗OS、神经炎症反应和神经兴奋性毒性反应中的作用和机制。TBI后,Ca2+超载、炎症介质释放和ROS过剩,导致颅内抗氧化机制和能量代谢失衡,BBB渗透性改变,进而产生一系列神经官能综合征。而这一级联反应也为预防sTBI导致的神经功能障碍提供了药物干预的窗口期。Sal作为一种天然、安全的抗氧化剂,在多种全身性疾病模型中显示出强大的抗氧化和神经保护能力。Sal通过消除多余ROS、调节线粒体稳态、抑制炎症因子释放和稳定细胞结构,有效减少神经细胞凋亡和炎症反应,促进神经细胞再生。然而,随着野生Sal的植物资源减少,传统提取方法难以满足市场需求。因此,化学合成和生物合成等方法已在当前研究中得到开发和利用。Sal在临床上多用于中风康复和治疗高原缺氧。虽然sTBI及其治疗的临床研究和动物研究较多,但大部分研究单一,实验成果有限,且尚无数据可以为患者提供安全有效的临床证据。另外,到目前为止,还没有一种Sal制剂进入医学临床试验。因此,为了更好地指导Sal的临床应用,应当结合Sal在分子生物学、遗传学和代谢工程中的广阔应用前景,采用先进的技术和方法对Sal进行更深入的研究,以期待更多高质量和大型的临床实验,为Sal在临床中应用提供进一步的依据。
