NLRP3 炎性小体及其下游细胞因子在哮喘中的作用研究进展
2024-05-06黄莎莎黄丽端许秋凤陈淑珍
黄莎莎 黄丽端 许秋凤 陈淑珍
支气管哮喘简称哮喘,是一种慢性疾病,表现为气道高反应性(airway hyperresponsiveness, AHR)、气道重塑、黏液分泌增多、喉中发出轰鸣声等[1]。哮喘是世界上仅次于癌症的第二大致死和致残疾病,严重危害人们的健康。哮喘发病过程有多种细胞参与,包括嗜酸性粒细胞(eosinophil, Eos)、中性粒细胞(neutrophil, Neu)、树突细胞(dendritic cell, DC)、T 细胞、肥大细胞等。
根据气道炎症模式不同,可将哮喘分为 2 型高炎症性哮喘和 2 型低炎症性哮喘。根据发病时痰液中显著增加的细胞不同,2 型高炎症性哮喘可分为嗜酸性粒细胞性哮喘(eosinophilic asthma, EA)和混合性哮喘,2 型低炎症性哮喘可分为中性粒细胞性哮喘(neutrophilic asthma, NA)和寡细胞性哮喘[1]。临床患者中以 EA 或 NA 患者居多。既往研究表明,核苷酸结合寡聚化结构域样受体家族含热蛋白结构域蛋白 3 (nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3, NLRP3)炎性小体贯穿于哮喘的各个时期,在其过度激活后,机体产生大量炎性细胞因子白细胞介素-1β (interleukin 1β, IL-1β)、IL-18 以及 IL-33,将导致机体出现气道重塑等不良后果,在哮喘的发生、发展中发挥重要作用。本文主要介绍 NLRP3 炎性小体及其下游细胞因子 IL-1β、IL-18、IL-33 在哮喘中的作用。
一、NLRP3 炎性小体
1.NLRP3炎性小体的结构: NLRP3是胞质内NOD样受体(NOD-like receptor, NLR)中的一员,在固有免疫反应中扮演重要角色。正常状态下,NLRP3 蛋白处于无活性状态;当与其配体结合时,中部结构域发生变构,招募接头蛋白ASC (apoptosis-associated speck-like protein containing a CARD, ASC)并与其结合,进而共同招募半胱氨酸天冬氨酸蛋白水解酶 1 (cysteinyl aspartate specific proteinase 1, caspase-1)前体pro-caspase-1,三者共同组成NLRP3 炎性小体[2]。
2.NLRP3炎性小体的激活机制: 目前认为激活 NLRP3炎性小体需要两种模式受体配合完成,其激活过程需要两个信号。首先,Toll样受体等识别病原体相关分子模式,活化核因子κB,诱导NLRP3、pro-IL-1β、pro-IL-18、pro-IL-33蛋白表达,此为启动信号;随后,胞质内模式受体NLRP3 识别危险信号并与ASC、pro-caspase-1组装成NLRP3炎性小体,使pro-caspase-1自切割为成熟的caspase-1。最后,pro-IL-1β、pro-IL-18、pro-IL-33被 caspase-1切割形成成熟的IL-1β、IL-18 和 IL-33,分泌至胞外,参与炎性反应,这是激活信号[2]。
NLRP3 炎性小体的激活信号错综复杂。研究显示,溶酶体损伤、细胞内外离子(如K+、Cl-、Ca2+等)浓度异常、活性氧激活、高尔基体解体以及高尔基体与线粒体相关内质网膜共同作用等状况都可能引起 NLRP3 炎性小体激活[3]。

图1 NLRP3 炎性小体激活信号通路
3.NLRP3炎性小体与哮喘: 作为固有免疫的重要组成部分,NLRP3 炎性小体是治疗哮喘的一个关键靶点。哮喘急性发作的患儿经布地奈德混悬液雾化吸入治疗后,哮喘症状明显缓解,这与IL-1β 水平、NLRP3 mRNA 的降低有关[4]。Ma等[5]研究发现,哮喘小鼠肺泡巨噬细胞中的NLRP3、caspase-1 和 IL-1β 表达升高;敲除NLRP3基因(NLRP3-/-)小鼠肺泡灌洗液(bronchoalveolar lavage fluid, BALF)中Eos和Neu数量减少、IgE浓度降低,AHR得到缓解。此外,NLRP3抑制剂RRx-001和MCC950,caspase-1抑制剂VX-765和 Ac-YVAD-CHO都能有效改善小鼠哮喘症状[5,6]。这些研究表明,NLRP3炎性小体及其下游产物caspase-1在哮喘的发病过程中发挥着巨大作用,但其作用机制仍有待于进一步研究。随着研究的不断深入,NLRP3 炎性小体下游细胞因子 IL-1β、IL-18 和 IL-33 也被证实与哮喘关系紧密,深入了解三者在哮喘中的作用,将为治疗哮喘提供新思路。
二、IL-1β、IL-18、IL-33 概述
1.IL-1β: IL-1β主要由骨髓细胞产生,其中,巨噬细胞是其最主要的来源。作为机体免疫的重要组成部分,巨噬细胞在过敏原等刺激下可以产生过量炎性细胞因子,引发或加重哮喘,给机体造成不可逆转的损伤[7]。caspase-1是使IL-1β切割成熟的关键酶,除了caspase-1,caspase-8、caspase-11以及组织蛋白酶 G等也可以切割pro-IL-1β并使其成熟,但效率远不如caspase-1[8]。
2.IL-18: IL-18主要在骨髓细胞、上皮细胞中表达,其中 Neu是哮喘患者血液中主要表达IL-18的白细胞。Neu活化后释放的炎性细胞因子、蛋白酶、氧自由基等物质将诱导机体产生AHR、气道重塑等不良症状[9]。IL-18与IL-1β同属于IL-1家族,caspase-1、caspase-8、颗粒酶B、蛋白酶3等也能切割活化IL-18[8]。
3.IL-33: IL-33也是IL-1家族中的一员,主要在上皮细胞、DC中表达。上皮细胞与 DC 在哮喘的发展中发挥重要作用,其中上皮细胞与气道重塑、AHR等有关,而DC对于诱发、维持和抑制肺部变应性炎症至关重要[7,10]。研究普遍认为,成熟的caspase-1切割pro-IL-33使其成熟并分泌至胞外,发挥生理活性。然而,另有研究者提出IL-33的生成仅受NLRP3蛋白影响,而非NLRP3炎性小体;还有研究认为成熟的caspase-1导致IL-33失活,与上述经典炎性小体激活途径调控IL-33活化的理论大相径庭[11,12]。同样,除了caspase-1,研究显示组织蛋白酶G等多种蛋白酶也能对IL-33进行切割活化[13]。
总之,尽管IL-1β、IL-18、IL-33可被多种酶切割成熟,但最主要的仍是通过经典NLRP3炎性小体激活途径,即caspase-1切割来完成。同时,表达三者的细胞,大多参与哮喘的发生、发展,并发挥着重要作用。深入研究NLRP3炎性小体下游细胞因子IL-1β、IL-18、IL-33将为治愈哮喘提供思路。
三、IL-1β、IL-18、IL-33与嗜酸性粒细胞性哮喘
EA属于2型高炎症性哮喘,是辅助性T淋巴细胞1 (T helper cell 1, Th1)/Th2细胞失衡而致的哮喘类型,初始CD4+T细胞倾向分化为Th2[1]。Th2可分泌多种Th2型细胞因子(如IL-4、IL-5、IL-13等),这些细胞因子皆在哮喘的诱发过程中发挥作用。在正常机体内,Eos只能生存2~5天;但在EA患者体内,Eos的寿命被Th2型细胞因子大大延长,使气道被大量Eos浸润,这是发生EA 的前提条件[14]。在哮喘患者中,EA患者占绝大多数,NLRP3炎性小体下游细胞因子在其发生、发展中发挥重要作用。
1.IL-1β: IL-1β可以招募Eos到气道中,通过增强Eos活性、抑制其凋亡并增加Th2型细胞因子分泌从而诱发哮喘[15,16]。IL-1受体拮抗剂(interleukin-1 receptor antagonist, IL-1Ra)是IL-1β的天然抑制剂,可与IL-1β结合,阻断IL-1β的作用。研究显示,IL-1Ra-/-小鼠AHR加重、炎性细胞因子分泌增加[17]。综上所述,IL-1β可促进哮喘的发生、发展,治疗靶点是治疗哮喘的一个重要思路。
2.IL-18: IL-18可提高IgE水平,促进Eos趋化因子eotaxin的产生与分泌,增加炎症部位Eos数量,且可以同时增强Th1、Th2型反应,产生对应的下游因子,如干扰素-γ (interferon γ, IFN-γ)、IL-13等引发哮喘[18,19]。使用IL-18中和抗体治疗后,小鼠的哮喘症状得到明显缓和[19]。然而,Hofstra等[20]用IL-18和IL-12联合治疗哮喘小鼠,检测治疗前后血清中IgE、BALF中Eos数量以及AHR程度,发现治疗后的小鼠各项体征都有所改善。研究指出,IL-18可与IL-12协同作用,共同促进Th1增殖与激活,诱导Th1介导的免疫反应,促进IFN-γ分泌,调节Th1/Th2细胞平衡,从而改善EA[20]。目前IL-18对EA的作用机制仍不完全清楚,无论是使其恶化或改善,都表明IL-18在EA中发挥着作用。
3.IL-33: IL-33可与肿瘤发生抑制蛋白 2 (suppression of tumorigenicity 2, ST2)结合,激活Eos等细胞,促进Th2型细胞因子分泌,加重哮喘症状,参与气道重塑,在哮喘、鼻炎等疾病过程中发挥重要作用[21,22]。An等[22]实验发现,ST2-/-小鼠明显降低了OVA致敏引发的AHR、炎性细胞浸润、肺组织中Th2型细胞因子、纤维化相关蛋白的表达以及血清总IgE 水平。冯净净等[21]研究发现,哮喘小鼠IL-33、IgE水平升高,BALF中Eos比例、Th2型细胞因子升高;使用IL-33抗体阻断后,IL-4、IL-13、IL-17的表达均下降。上述研究表明,调控IL-33水平有望成为治疗哮喘的有效方法。
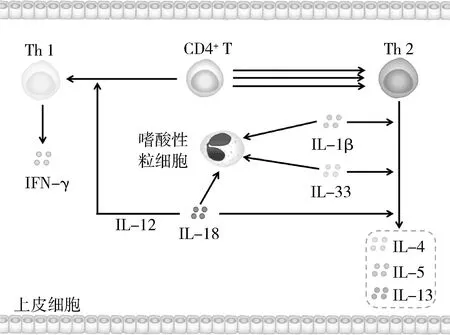
图2 IL-1β、IL-18、IL-33在嗜酸性粒细胞性哮喘中的作用机制
四、IL-1β、IL-18、IL-33与中性粒细胞性哮喘
NA有别于传统的EA,是较难治疗的哮喘类型,气道炎症程度高于EA,且对激素治疗效果不明显。NA属于2型低炎症性哮喘,是由 Th17/调节性T细胞(regulatory T cell, Treg)失衡诱导的,初始CD4+T细胞倾向分化为Th17[1,23]。Th17分泌的IL-17可以刺激巨噬细胞、气道上皮细胞、成纤维细胞等产生IL-8、IL-1β等多种细胞因子,其中IL-8又被称为中性粒细胞因子,可以激活和招募Neu,诱导AHR及促进气道重塑[23]。因此,IL-17、Th17已成为治疗NA的热门靶点。与此同时,NLRP3炎性小体的下游细胞因子也在其中发挥着重要作用,值得进一步探究。
1.IL-1β: IL-1β能促进粒细胞集落刺激因子(granulocyte colony stimulating factor, G-CSF)分化Neu,能诱导IL-8合成,还能促进Th17分化进而影响IL-17产生,直接或间接诱发中性粒细胞性炎症,导致NA的发生[8,15,24]。IL-1β需与IL-1受体(interleukin-1 receptor, IL-1R) 结合后才能进行信号转导,发挥功能。研究显示,IL-1R-/-哮喘小鼠的气道炎症能被有效缓解[15]。综上所述,IL-1β贯穿NA发生、发展的过程,是影响NA的重要因子,深入研究IL-1β的作用将为治疗NA提供新思路。
2.IL-18: IL-18可促进Neu增殖并将其招募至气道,诱导NA发生[9]。IL-18还能单独或协同IL-23、IL-12促使Th17分化和IL-17生成[25,26]。在难治性哮喘小鼠模型中,IL-18-/-小鼠的BALF中Neu减少,且AHR程度也有所降低,气道状态更好。另有实验证明,在原代人类肺泡上皮细胞中,IL-18可以诱导IL-6、IL-8表达,促进Th17发育,招募Neu至炎症部位[27]。综上所述,IL-18可单独或联合IL-23、IL-12促进NA发生,减少或抑制IL-18可以缓解和治疗哮喘。
3.IL-33: IL-33可直接作用于肥大细胞,促进IL-1β和IL-6产生,从而增强Th17分化,最终可能诱发Neu主导的气道炎症。IL-33还可通过促进DC成熟抑制Treg分化,诱导Th17生成,升高IL-17水平。Park等[28]研究显示,在IL-33的刺激下,未成熟的DC (IM-DC)将转化为成熟的DC (IL-33-MAT-DC)。IL-33-MAT-DC可以通过减少Treg转录调节因子 Foxp3的表达来抑制Treg的分化。IL-33在NA中的作用机制尚不明确,基于其对Th17的调控作用,IL-33有待于成为哮喘治疗的新靶点。
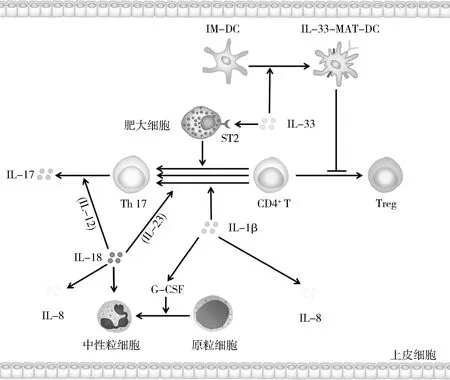
图3 IL-1β、IL-18、IL-33在中性粒细胞性哮喘中的作用机制
五、展 望
哮喘作为仅次于癌症的世界第二大致死和致残疾病,给患者带来了不可逆转的伤害。NLRP3炎性小体及其下游细胞因子IL-1β、IL-18和IL-33是影响哮喘发生、发展的关键。但目前为止,以其为治疗靶点的临床应用实例较少,它们在哮喘中的作用也尚未完全研究透彻。深入研究 NLRP3 炎性小体及其下游细胞因子在哮喘中的作用及机制,以便有的放矢地进行药物研究,为临床缓解和治疗哮喘提供思路。
利益冲突声明:所有作者均声明不存在利益冲突。
