MLST技术分析中国烟草野火病菌遗传多样性
2024-04-23林凡力彭建斐邓泽征郭璐璐姚廷山马皓月马冠华
林凡力,彭建斐,邓泽征,郭璐璐,姚廷山,马皓月,马冠华
(1西南大学植物保护学院,重庆 400716;2湖南省烟草公司怀化市公司,湖南怀化 418000)
0 引言
烟草野火病(Tobacco Wildfire)是细菌性叶部病害,由丁香假单胞杆菌烟草致病变种Pseudomonas syringaepv.tabaci引起,多集中于烟草幼苗阶段与大田阶段发病。该病害初发生时为水浸小斑点,外缘有黄色晕圈,温湿度适宜时形成无规则的褐色轮纹斑,后期病斑相连成片,形成近圆形或无规则大病斑;该病害可导致烟草烟叶大面积死亡或腐烂,仿佛火灼烧状。WOLF 等[1]1917 年首次报道该病害在美国烟区发生,之后各国陆续有报道。20 世纪中期该病害在中国就有零星发生,到80年代中后期已在多个省(区)大流行,并从次要病害上升为主要病害,尤其在四川、云南等产烟区,该病害连年发生且为害严重[2-6]。中国对烟草野火病研究多集中于病害流行学[7]、病菌致病性[8-9]、毒素及抗药性[10-12]等方面。该病原菌存在生理分化现象,主要表现为对长花烟与黄花烟的致病性差异。云南[13]、黑龙江和吉林三省[14-15]报道过烟草野火病菌遗传多样性研究,分子标记是研究真菌遗传多样性和种群结构的重要手段,可有效研究病菌与地理环境或寄主之间的关联。多位点序列分型(MLST)技术是近年流行的基于核酸序列测定细菌分型的方法,具有重复性强,分辨率高等优点,是一种更准确的分型系统方法,多应用于人体病原细菌研究,可比对分析各菌株间显著保守性表现的管家基因碱基分布改变。明晰病菌的遗传多样性可揭示同种病菌不同区系之间的遗传和进化,了解和掌握病菌遗传结构和多样性水平可为有效防治措施制定和品种选育提供强力支撑,为病菌的生物学特性研究提供新视角[13]。因此,笔者收集了湖南、重庆等5 个省(市)烟草野火病菌,采用多位点序列分型技术研究烟草野火病菌菌株之间的遗传多样性,以期为该病害的有效防控提供参考。
1 材料与方法
1.1 材料
从湖南、重庆、四川、黑龙江、吉林等5个烟草种植省(市)采集烟草野火病样,分离野火病菌。供试烟草品种为‘云烟87’,玉溪中烟种子有限责任公司生产。
1.2 方法
1.2.1 病原菌分离及致病性测定病样采用组织分离法分离病原菌[16],纯化保存备用。‘云烟87’烟草种子播种于灭菌土壤中,培育至3片真叶苗,移栽至无菌蛭石营养钵,放置于25℃温室光照培养,在烟苗5~7 片真叶期,使用注射器在叶片背面接种3.0×108cfu/mL 待测病菌菌悬液,完成柯赫氏法则证病。
1.2.2 病原菌分子鉴定按细菌DNA 快速抽提试剂盒说明[17],提取烟草野火病菌DNA,PCR 用细菌16srDNA常 规 引 物27F/1492R(27F: 5'-AGAGTTTGATCCTGGCTCAG- 3',1492R: 5'-TACGGYTACCTT GTTACGACTT-3'[14])。总反应体积为50 µL:ddH2O 37 µL,10×PCR buff 5 µL,2.5 mol/L dNTPs 4 µL,10 µmol/L Primer (+) 1 µL,10 µmol/L Primer(-)(10µmol/L)1µL,5 U/µLTaqDNA聚合酶1µL,30 ng/μL DNA模板1µL。
PCR反应程序:95℃,预变性5 min;每一个循环为94℃变性0.5 min,60℃,退火1 min,72℃延伸1 min,共30个循环,最后72℃延伸10 min,PCR放置产物4℃冰箱储存备用。
扩增产物送华大基因科技股份有限公司测序,应用DNA Star软件得到16s-rDNA序列,提交至NCBI比对,进行病原菌分子鉴定。
1.2.3 病菌遗传多样性 从NCBI 网站下载JWJF01000000 野火病菌菌株的基因组序列,找到11个管家基因的序列[18],提交上海生工生物工程有限公司使用primer 5.0软件设计引物,PCR筛选较优管家基因及退火温度,最终优选3 个管家基因(表1),进行MLST分析。

表1 JWJF01000000野火病菌菌株基因组中7个管家基因的功能和引物序列
DNA提交华大基因科技股份有限公司测序后,运用DNA Star双向拼接并校对。应用DANMAN软件多序列比对管家基因PCR 产物的若干碱基序列,确定“菌株的序列型(ST)”;登陆http://www.eBURST.mlst.net/,应用eBURST V3 软件,以Gap、Pgi及Pfk基因标准,绘制供试菌株种群遗传关系图谱。
2 结果与分析
2.1 病原菌的分离与鉴定
共采集病样112 份,分离获得疑似烟草野火病菌菌株160株,菌株来源分别为四川40株,湖南20株,重庆40 株,吉林40 株,黑龙江20。按照柯赫氏法则验证,其中144 株为烟草野火病菌Pseudomonassyringaepv.tabaci。
2.2 病原菌分子鉴定
DNA快速抽提试剂盒提取144株菌株DNA,选择细菌通用引物27F/1492R,PCR 扩增后经琼脂糖凝胶电泳,得到1 条约为600 bp 清晰条带。测序拼接得到该部分的基因序列,序列大小为589 bp(见图1)。

图1 部分烟草野火病病原菌基因组DNA的16s-rDNA PCR扩增
BLAST分析结果表明,144株菌株序列P.syringae(CP000075.1)和P.amygdale(CP020351.1)相似性达到99%以上,结合致病性检测结果,分离获得的144株菌株鉴定为烟草野火病菌P.syringaepv.tabaci。
2.3 多位点序列分型
2.3.1Gap基因eBURST V3 分析结果表明(参照Gap基因标准),待测烟草野火病菌株均属于同一亚群以及ST85 这一单独群。除了单独的29 个ST,待测菌株构建4 个CC,即CCl(77 个ST 组成)、CC2(10 个ST 组成)、CC3(3 个ST 组成)和CC4(2 个ST 组成)(见图2)。
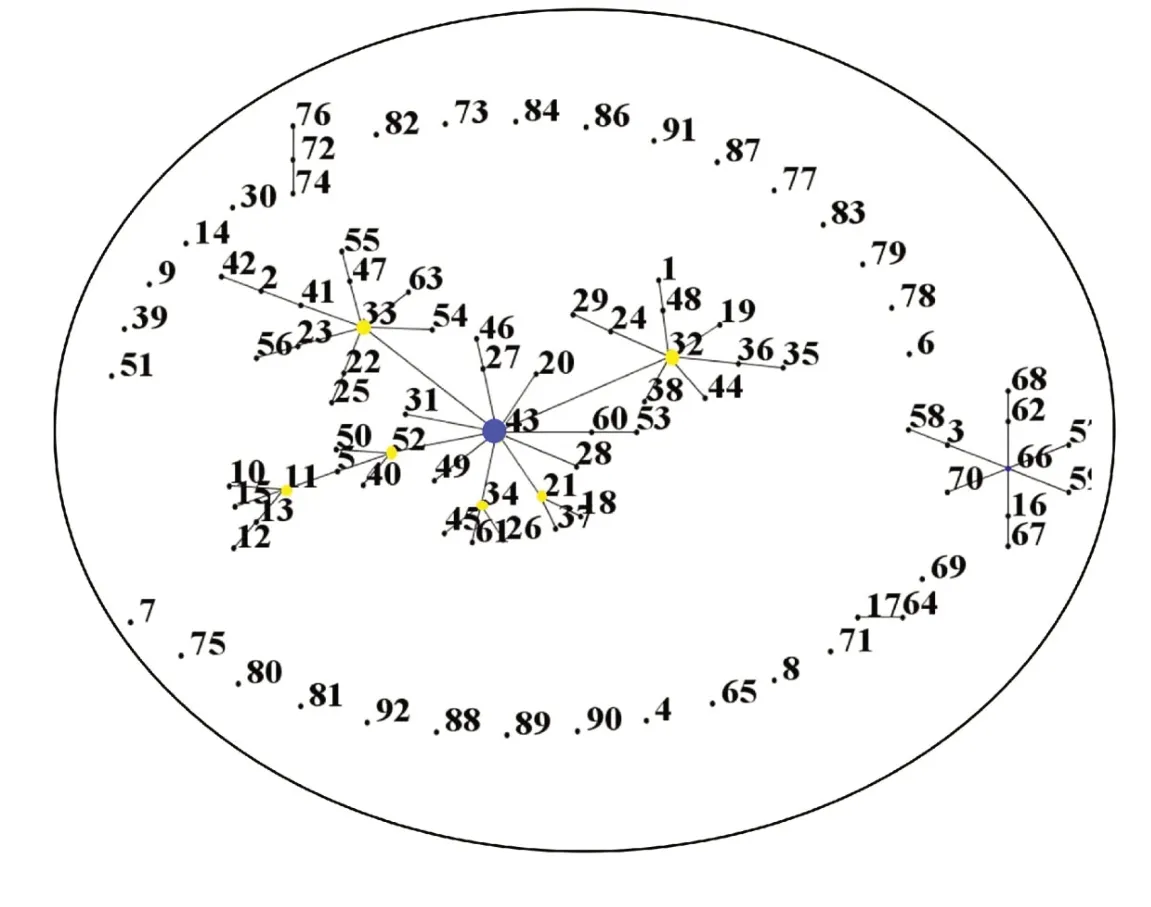
图2 共享Gap基因标准下烟草野火病菌种群结构
2.3.2Pgi基因eBURST V3分析结果表明(参照Pgi基因标准),待测烟草野火病菌株归至亚群1~亚群5及4个单一群。亚群1包含58个ST,核心型为ST43,菌株11 株,其中吉林5 株,四川4 株,湖南2 株;亚群2 包含18 个ST,核心型为ST66,菌株5 株,其中重庆3 株,四川2株;亚群3包含4个ST;亚群4包含4个ST,均来自黑龙江;亚群5 包含2 个ST,均来自四川。4 个单一群菌株则分别来自湖南、吉林、重庆与四川。
亚群1 中的ST 构建出CCl、CC2 这2 个同源复合体(除去ST9、ST14、ST30、ST39、ST51、ST73、ST82、ST84),其中CC2 含有3 个ST,分别为ST72、ST74 和ST76。亚群2 中的ST 形成2 个CCl、CC2(除去ST4、ST6、ST8、ST65、ST69、ST71),CC2 含有2 个ST,分别为ST17和ST64(见图3)。

图3 共享Pgi基因标准下烟草野火病菌种群结构
2.3.3Pfk基因eBURST V3分析表明(参照Pfk基因标准),供试菌株聚集于亚群1、亚群2、亚群3、亚群4 及30 个单一群。亚群1 为CCl、亚群2 为CC2、亚群3 为CC3,而ST6、ST7、ST8、ST9等均为单一群(图4)。

图4 共享Pfk基因标准下烟草野火病菌种群结构
综合分析结果,基于Gap基因标准,供试烟草野火病菌菌株仅1 个亚群与单一群,菌株间的多态性以及差异性较小;基于Pfk基因标准,由于过多单一群,亚群和单一群间的遗传差异性也较小;而基于Pgi基因标准的分型较好,ST间的遗传性差异较大,分为优势亚群1和亚群2。亚群1中核心ST型ST43为中国主要种群,来自四川、湖南、吉林。亚群2中核心ST型ST66来自四川与重庆。同一核心型菌株间不存在显著相关性。
2.4 烟草野火病病菌分型关联分析
对中国东北和南部片区采集病样分离纯化鉴定的烟草野火病菌进行亚群划分分析,结果表明南北方烟草野火病菌ST 型的差异不明显,而亚群4 较为特殊,其包含的菌株均来自东北,推测这些菌株由同种ST型发展蔓延而来。试验采集的病样来自西南地区主栽品种云烟87,东北地区主栽品种吉烟9 号和龙江911,分析发现,亚群1 和亚群2 的核心ST 型包含的烟草品种不同,相同ST型可能来自不同的烟草品种,而不同ST型也可能来自同一烟草品种(表2),推测烟草品种与病菌组群间无关联性。

表2 烟草野火病菌的MLST分型关联分析
3 结论与讨论
中国主栽烟草品种多数不抗烟草野火病,该病防控较困难[18-19]。目前,选育优质抗野火病品种是防治该病害最有效的方法,而明确其病菌遗传多样性是抗病品种选育的前提[20-23]。中国各地烟草野火病菌ST型不同,同一ST 型可能会诱导病害大面积发生,目前因环境、人为因素等的影响,已进化发展为不同的ST型[24]。
本试验中,烟草野火病菌遗传多样性分析的144株菌株,来自四川、重庆、湖南、吉林及黑龙江5省(市)的3 个烟草品种,地理跨度较大。Pgi基因标准条件下,供试菌株分为5个亚群和4个单一群,其中,亚群1内的ST 型数量最多,其核心型菌株来源3 个省,菌株分布较广,不存在区域优势;亚群2 内的ST 型数量次之,其核心型菌株来自两个省,各菌株关系松散,不在同一CC上;亚群5内的ST型数量最少,菌株皆来自四川;而4 个单一群菌株分别来自吉林、湖南、重庆与四川4个省(市),结果表明,烟草野火病菌组群划分同烟草品种不存在关联。
以烟草野火病菌来源的地域分析,试验所用的供试菌株主要来自中国东北与西南,地理跨度较大,结果表明,优势亚群1 和亚群2 囊括了来自吉林、湖南、重庆、四川及黑龙江5 个区域的菌株,菌株ST 型无明显差异。亚群分析结果表明,亚群1的核心型ST来自吉林、四川、湖南,说明亚群1所包含的菌株分布较广,不存在区域优势;亚群2 核心型ST 源于四川和重庆,但亚群内各菌株关系松散,不在同一CC 上;亚群4 的所有ST型菌株均来自东北。
烟草品种来看,亚群1 和亚群2 的核心ST 型来源的烟草品种不同,表明相同ST型可来自不同的烟草品种,不同ST 型也可能存在于相同烟草品种,说明烟草野火病菌组群划分同烟草品种不存在相关性。陈赟娟等[25]结果表明,烟草野火病菌群体致病性存在差异,来源地也不同,两者之间无关联性。刘雅婷等[13]发现,云南烟草野火病菌群体遗传多样性突出。夏纬跃等[26]发现,吉林省辖区内70株烟草野火病菌在区域上的遗传多态性丰富。本试验研究结果与之类似,供试烟草野火病菌存在丰富的遗传多样性。
