阿蓬江黔江段干支流水体微生物群落特征分析
2024-04-22许田甜田茂强刘朝红李小林胡廷英
许田甜,田茂强,刘朝红,李小林,胡廷英
[1.重庆新阳地理信息有限公司,重庆 401147;2.自然资源部重庆典型矿区生态修复野外科学观测研究站(重庆地质矿产研究院),重庆 401120;3.长江师范学院绿色智慧环境学院,重庆涪陵 408100]
我国水体微生物研究主要集中在长江[1]、黄河[2]等主要河流地,以及鄱阳湖[3]等典型湖泊,对于支流等小型河流、湖泊及其他小型水域的微生物缺乏研究。河流生态系统中,浮游细菌是重要组成部分,在水体中浮游细菌数量庞大,代谢活动强烈,是高级生物的主要食物,对水中污染物的降解和水循环过程有重要作用[4]。在水生态系统中,微生物对水体外的环境因子的变化十分敏感。近年来,众多学者研究了浮游细菌与环境因子之间的相关性[5],但大都集中在海洋和湖泊[6]。河流作为一个动态系统,它们的环境条件在时间和空间尺度上具有高度差异性[7]。相关研究[8]表明其在湿地、养殖废水和深海沉积物等环境中优势类群均是变形菌门,且大多数在生物脱氮除磷和污染物降解过程中起重要作用的微生物也是变形菌门[9]。在纲水平上,β-变形菌纲占据的比例最大,达到了11.01%到31.50%不等,其大多数存在于河流和湖泊中(松花江[10]、海宁长山河[11]、东江[12]、Prealpine 湖[13]等),对氮、磷等污染物的降解起到了重要的作用[14]。
本研究选择阿蓬江(黔江段)为研究区域,对其水体中的微生物种类、多样性和群落分布特征进行分析,以期为观察阿蓬江水污染状况,为长江上游水环境保护和长江流域微生物群落特征和多样性研究提供数据支持。
1 材料与方法
1.1 研究区概况及样品采集
阿蓬江(黔江段)位于重庆市黔江区南部,全长约60 km。主要支流有太极河、细沙河、段溪河及黔江河等河流。于2019年在阿蓬江(黔江段)干支流中设定11 个样点并取样,其中支流设有4 个样点,分别是QW1、QW7、QW9、QW11 样点,其余7 个全是干流样点。
1.2 水体微生物群落结构分析
1.2.1 水体微生物总DNA提取
水样通过0.22 μm 的滤膜进行抽滤,过滤好后的滤膜放置在无菌离心管中于-20 ℃保存,并使用15 kg干冰送测。
1.2.2 水体微生物高通量DNA测序
用特定的DNA 提取试剂盒进行基因组DNA 提取后,再用0.8%琼脂糖凝胶电泳检测DNA。16S 引物使用:341F(5'-CCTAYGGGRBGCASCAG-3') 和806R(5'-GGACTACHVGGGTWTCTAAT-3')以稀释后的基因组DNA 为模板,根据测序区域的选择,使用带Barcode 的特异引物进行PCR。每一个25 μL 的体系包括1×PCR buffer、1.5 mM MgCl2、0.4 μM dNTPs、正向和反向引物各1.0 μM、0.5 U KOD-Plus-Neo 酶(TOYOBO) 和10 ng 模板。PCR 程序包括起始94 ℃1 min,然后30 循环(变性94 ℃20 s,退火54 ℃30 s 和 延伸72 ℃30 s),最后72 ℃5 min。每个样本进行3 个PCR 技术重复。PCR 与1/6 体积的6×loading buffer 混合,用2% 琼脂糖凝胶电泳检测。取目的条带用来回收,回收使用QIAquick Gel Extraction Kit(QIAGEN)。用 Qubit@ 2.0 Fluorometer (Thermo Scientific)定量。最后等摩尔量混合。建库使用TruSeq DNA PCR-Free Sample Prep Kit,构建好的文库经过定量和文库检测合格后,使用罗宁生物的Miseq 平台PE300 模式测序。测序工作在上海罗宁生物有限公司进行。
1.3 数据分析与处理
高通量测序产出的原始图像数据文件经CASAVA软件进行碱基识别后转化为原始的测序序列,称为Raw Data 或Raw Reads,结果以Fastq 格式储存。对序列进行质控,得到高质量序列,质控后得到的序列称为Clean Tag。使用UCHIME7 算法去除嵌合体,得到Effective Tag。为了方便研究样本的物种组成及多样性信息,对序列进行聚类产生操作时的分类单元,即OTU(Operational Taxonomic Units)。在97%的相似水平下进行OTU 聚类分析,每个OTU 代表的是一类相似序列的集合,通过OTU 聚类分析,得到OTU 丰度表,此丰度表是后续分析的基础文件。为防止测序深度对后续多样性分析产生影响,对数据进行重抽样,使得各样本保持一致的序列数。用Circos 网站在线绘图功能(http://circos.ca/)对阿蓬江水体微生物种群门、纲、目、科水平的相对丰度变化情况进行可视化表达。
2 结果与分析
2.1 阿蓬江干支流水体微生物组成成分
在阿蓬江(黔江段)区域所取样的11个样点所得的所有细菌16 SrDNA 片段中,将得到的序列按门、纲、目、科分类,可以知道研究区域所取样的11个样点共有27门、46纲、98目、158科。
2.2 阿蓬江干支流水体微生物群落多样性
通过采用α 多样性分析,对阿蓬江干支流水体微生物群落多样性进行表征,包括物种丰富度指数(Chao 1)、Shannon 多样性指数(H’)和Simpson 多样性指数(D)(见表1)。对阿蓬江干支流水体样品测序后,获得97%相似水平及以上的OTUs分别有约360个和337 个,且对微生物群落覆盖率达到99%,OTU值干流明显大于支流。Chao 1 丰富度指数表现为干流水体大于支流水体,干支流微生物群落OTU 值均低于Chao 1丰富度指数,Shannon 和Simpson 指数也均表现为干流水体大于支流水体。总体而言,阿蓬江水体微生物的群落丰富度和多样性均表现为干流大于支流。

表1 阿蓬江(黔江段)水体微生物群落多样性指数分析
2.3 门水平上的微生物群落分布特征
通过高通量测序分析,发现细菌群落在门水平上有27个细菌门,其中主要为变形菌门、放线菌门和拟杆菌门等细菌门,而在这些细菌门中占据绝对优势的是变形菌门,说明阿蓬江(黔江段)是以浮游细菌组成的典型的淡水水体,符合我国典型的淡水水体微生物组成的群落特征。
从表2 可知,在干流中的优势细菌门是放线菌门、拟杆菌门、厚壁菌门、浮霉菌门和装甲菌门,支流中的优势细菌门则是变形菌门、疣微菌门、异常球菌门、芽单胞菌门和奇古菌门,从标准偏差来看,放线菌门、拟杆菌门和异常球菌门在干支流的优劣势并不明显,拟杆菌门和厚壁菌门虽然在占比上是干流优于支流,但是其干流的标准偏差是远大于支流的,相反的是,异常球菌门在占比上是支流优于干流,其标准偏差是支流略大于干流,由此可看出细菌群落在空间的分布上是有一定的空间差异,但其波动幅度不大。
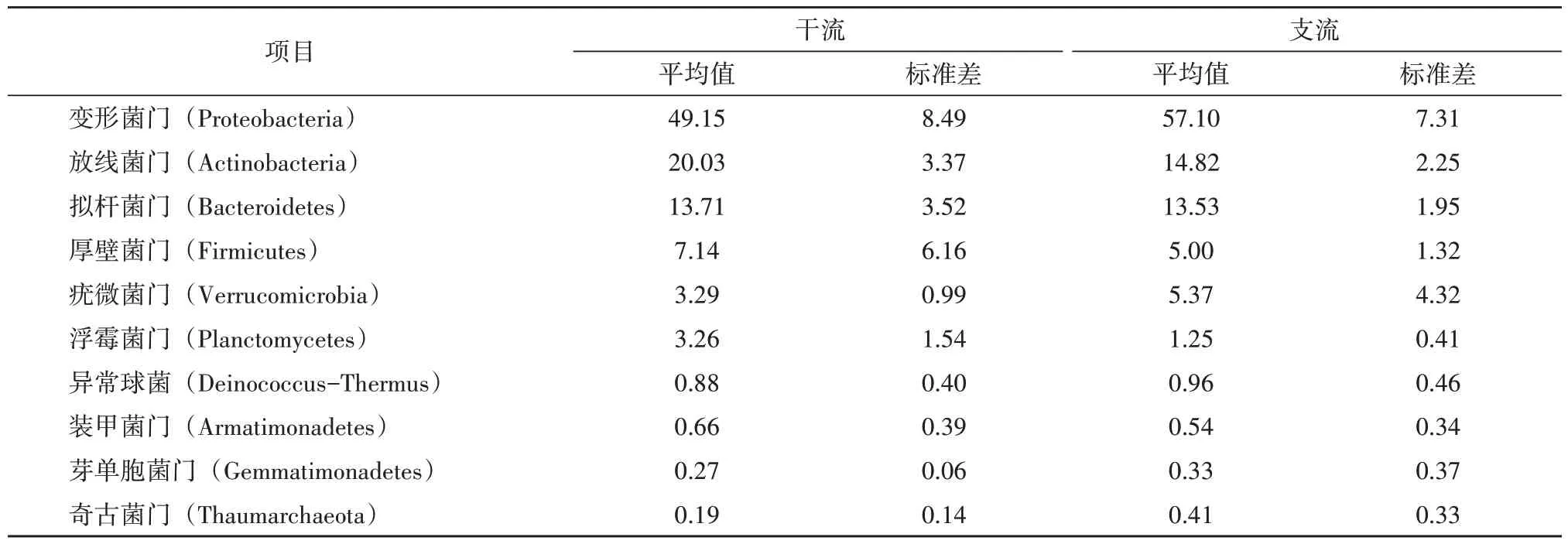
表2 门水平上干支流水体前10位微生物占比 单位:/%
2.4 纲水平上的微生物群落结构分析
通过高通量测序分析,在纲水平上一共发现46个细菌纲。从表3 的干支流占比分析来看,干流中的优势细菌纲有放线菌纲和α-变形菌纲,在支流中占据优势的细菌纲是γ-变形菌纲和拟杆菌纲。从标准差来看,首先γ-变形菌纲在支流中的标准差是要小于干流,其次是放线菌纲、拟杆菌纲和α-变形菌纲,其标准差均是干流大于支流,最后是疣微菌纲和Acidimicrobiia,这两大细菌纲在干支流中的标准差相距太大,疣微菌纲在支流中的标准差是干流中的4倍,Acidimicrobiia 在干流的标准差是支流的3 倍,所以这两大细菌纲在干支流中的空间分布有一定的波动幅度。
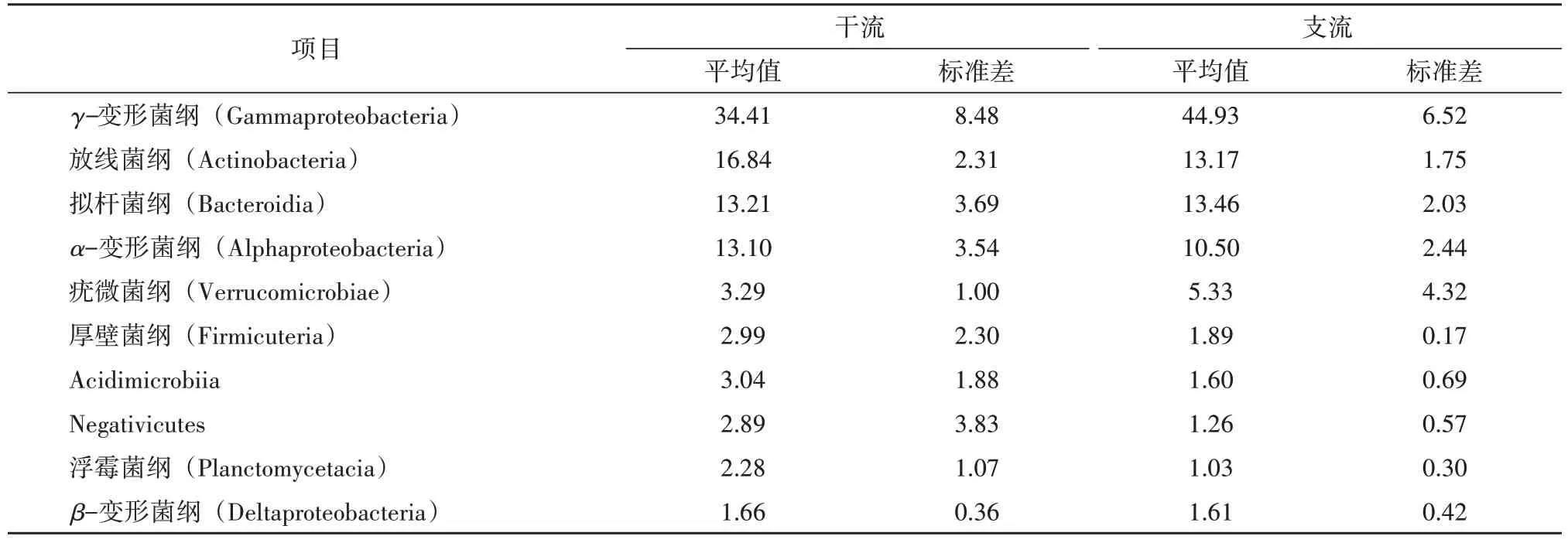
表3 纲水平上干支流水体前10位微生物占比 单位:%
2.5 目水平上的微生物群落结构分析
目水平上,一共发现98 个细菌目,如表4 所示,最优势的细菌目是伯克氏菌目,从干支流占比对比分析,伯克氏菌目在支流中的比例大于干流中所占比例,法兰克氏菌目干流中所占比例大于支流,从两者标准差来看,干支流中的空间分布没有明显差异。拟杆菌目和鞘脂单胞菌目在干流中的占比略优于支流,而其在干流中的空间分布波动相比于支流是比较大的,而剩下的假单胞菌目、噬几丁质菌目、热纤梭菌目等细菌目的相对比例差距不大,空间波动幅度不明显。

表4 目水平上干支流水体前10位微生物占比 单位:%
2.6 科水平上的微生物群落结构分析
科水平上,细菌科较多,所有样品中一共有158个细菌科,如表5 所示,最优势的细菌科是伯克氏菌科。从干支流的占比来看,支流中占比较大的是伯克氏菌科,而在干流中所占比例较大的是鱼孢菌科。从标准差的对比来看,伯克氏菌科和鞘脂单胞菌科在干流中的空间波动幅度远大于支流,鱼孢菌科、红杆菌科和假单胞菌科在干支流中的空间分布没有明显差异,而未列出的细菌科占据比例较大,在空间分布上也没有明显差异。

表5 科水平上干支流水体前10位微生物占比 单位:/%
3 讨论与结论
3.1 讨论
利用Illumina测序技术的16S扩增子测序,检测了阿蓬江(黔江段)水体微生物群落的组成成分,发现所取样的11 个样点共有27 门、46 纲、98 目、158 科。在门分类水平上,变形菌门是11个样品中最优势细菌门,如吴晓冰等[15]在研究长江干流浮游细菌群落结构中变形菌门(7.49%~85.63%)和张旭[16]在研究海宁市不同类型区域河流底泥微生物群落结构特征研究时,其中变形菌门为细菌门中最优势类群,均符合典型的淡水水体微生物的组成特征。变形菌门在研究区域呈现出支流含量高、干流含量低的特征,变形菌门在所有样点中都是绝对优势类群,说明变形菌门相比其他细菌更具有适应性;放线菌门和拟杆菌门作为次优势类群,在研究区域呈现出干流含量高、支流含量低的特征;而剩下的厚壁菌门、疣微菌门及浮霉菌门等细菌门的占比相对较低,在10%以下。在纲水平上,γ-变形菌纲是最优势类群,在研究区域呈现出支流含量高、干流含量低的特征;其次次优势类群是放线菌纲、拟杆菌纲及α-变形菌纲,在研究区域呈现出干流含量高、支流含量低的特征;其余的疣微菌纲、厚壁菌纲及β-变形菌纲等细菌纲的占比均不超过10%。γ-变形菌纲、放线菌纲、拟杆菌纲及α-变形菌纲在研究区域是优势类群,这与我国大多数淡水生态系统的调查结果是一样的[17-19]。在目和科水平中伯克氏菌均处于优势地位,其干支流的占比差距明显。初步分析伯克氏菌在支流上含量高的原因可能是支流采样点都是河流,其周围城镇发展迅速,其发展产生的各种污水导致水体污染加剧,其水体内营养物质增多,细菌含量增高。
3.2 结论
1)通过微生物16S片段高通量测序分析,阿蓬江(黔江段)所取得的11 样品在界的水平上总共发现27门、46纲、98目、158科。
2)阿蓬江水体微生物的群落丰富度和多样性均表现为干流大于支流。
3)在门水平分类上,以变形菌门为绝对优势类群,在纲水平分类上则是以γ-变形菌纲和放线菌纲为优势类群,在目分类上以伯克氏菌目为优势类群,在科分类上以伯克氏菌科占据绝对优势。
4)基于高通量测序数据分析,干支流空间差异较为明显,在门水平上,拟杆菌门和厚壁菌门在干流中的空间分布波动幅度较大,疣微菌门和芽单胞菌门在支流中的空间分布有较大差异。纲水平上,疣微菌纲在支流的标准差是干流的4 倍,Acidimicrobiia 在干流的标准差是支流的3 倍,所以这两大细菌纲在干支流中的空间分布有一定的差异。在目分类上,细菌目的空间分布有一定的差异,但热纤梭菌目、Microtrichales 和Selenomonadales 在分布上差异较大。在科分类上,细菌科的空间分布没有明显的差异,其中的鞘脂单胞菌科、Ilumatobacteraceae 和氨基酸球菌科的空间分布有些波动。
