一例多钒酸盐杂化材料的制备及高效催化烯烃环氧化
2024-04-17杜思宇王雪怡关致敏马宏芳
李 宁 杜思宇 王雪怡 杨 辉 周 涛 关致敏 费 鹏 马宏芳 蒋 尚
(山西大同大学化学与化工学院,大同 037009)
0 Introduction
The inorganic - organic hybrid materials, which combine inorganic and organic constituents, have been studied and have a broad scope of applications in catalysis, magnetism, photochemistry, and biomedicine[1-3].Thus, the preparation of functionalized inorganicorganic hybrid materials is of great significance. The key procedure in the preparation of such combined materials is the selection of eligible organic and inorganic constituents. Polyoxometalates (POMs) are a fascinating class of inorganic cluster materials possessing enormous structures and multifaceted applications.Because of their excellent performances, POMs have been researched extensively as precursors for the construction of hybrid materials[4-7]. Polyoxovanadates(POVs), as a special subclass of POMs, have aroused great interest owing to their electromagnetism, redox activity, and medical chemistry[8-10]. Additionally, the V—O fragments possess the potency to bind transition metals to form functional hybrids using their terminal oxygen atoms. These hybrid materials have been considered as oxidation catalysts to catalyze a variety of organic substrates. The materials not merely overcome the troubles of the easy aggregation and hard recovery of POMs but also enhance the stabilization and recyclability of catalysts[11-13].
Olefins epoxidation is a kind of important industrial catalytic reaction, and its epoxidation products are a kind of important organic intermediates, that have important applications in the fine chemical industry,petrochemical industry, polymer materials, and pharmaceutical synthesis[14-16]. In recent years, green chemistry has attracted great attention from researchers,hydrogen peroxide has high reactive oxygen content and its product is water, therefore, the reaction system using hydrogen peroxide as an oxidant has been widely studied. For the past few decades, varieties of POMs have been generally used as effective catalysts for the epoxidation of olefins. Up to now, many inorganicorganic hybrid POV materials have been applied to the selective oxidation of sulfides and alcohols and have shown efficient catalytic performance[17-20]. However,the investigation of hybrid POVs for olefin epoxidation catalysis is still rare[21-23].In addition,based on previous literature reports, cobalt-containing compounds show excellent catalytic effect and high selectivity of epoxidation products in various olefin epoxidation reactions and are potential epoxidation catalysts[24-26]. The combination of POVs and Co-complex may cooperatively interact giving rise to synergistic effects to enhance the catalytic activity.
On considerations of the above content, to investigate the oxidation catalytic performance of POVs, we successfully synthesized an inorganic-organic hybrid cobalt vanadate, [Co(pIM)V2O6] (1) (pIM=2-(2-Pyridyl)imidazole), by reacting CoCl2·6H2O with NaVO3and pIM under hydrothermal conditions. The compound exhibited the 2D network composed of VO4tetrahedra and CoO3N2square pyramid via both edge- and cornersharing. As a catalyst for epoxidation, the conditions of epoxidation of olefin were optimized,and the reusability of the catalyst was also studied.
1 Experimental
1.1 Materials and methods
The chemicals for the experiments were commercially sourced and no additional purifying was performed. Elemental analyses of Co and V were confirmed by PLASMASPEC (I)ICP atomic emission spectrometer, and the contents of C, H, and N were analyzed by a PerkinElmer 2400 CHN elemental analyzer.Powder X-ray diffraction (PXRD) was implemented on a Rigaku D/MAX-3 instrument and the radiation of CuKα(λ=0.154 2 nm) at 298 K and X-ray 40 kV/30 mA over a 2θrange of 5°-50°. The Fourier transform infrared (IR) spectra were collected using KBr pellets on an Alpha Centaurt FTIR system, implementing from 4 000 cm-1to 400 cm-1. Thermogravimetric (TG) analysis was determined with the Perkin-Elmer TGA7 apparatus with a heating speed of 10 ℃·min-1in an atmosphere of N2. The catalytic reaction process was monitored and evaluated by the GC-2014 (Shimadzu) system with biphenyl as an internal standard substrate.The collection of magnetic susceptibility data was used a SQUID magnetometer (Quantum Design, MPMS-5)with an external magnetic field of 1 000 Oe and a temperature region of 2 to 300 K.
1.2 Synthesis of compound 1
CoCl2·6H2O (0.24 g, 1.0 mmol), NaVO3(0.12 g,1.0 mmol) and pIM (0.15 g, 1.0 mmol) were added to 10 mL distilled water, and the reaction solution was adjusted to pH 4.2 with 1 mol·L-1HCl in the stirring process. The reaction solution was stirred for 15 min and then transferred to a 23 mL stainless reactor. The stainless reactor was placed in the oven at 170 ℃for three days and then decreased to ambient temperature at a rate of 10 ℃·h-1.Blocky crystals were collected by filtration, washing, and dried at ambient temperatures.Yield: 22.2% (V-based). Anal. Calcd.(%) for C8H7N3O6CoV2:C 24.3;H 1.7;N 10.6;Co 14.9;V 25.8;Found(%):C 23.9;H 1.8;N 10.9;Co 15.4;V 25.2.
1.3 X-ray crystallography
A regular block single crystal was selected to be wrapped with vaseline and encapsulated in a fine glass tube of appropriate size. Crystal data were obtained using a Bruker Smart-CCD diffractometer with monochromated MoKαradiation (λ=0.071 07 nm) at room temperature. Structure determination was fulfilled by direct methods using the SHELXS-2014 crystallographic program via the Olex 2 platform[27], and succedent atom refinement was accomplished using full-matrix least-squares procedures. In the process of refinement,all the non-hydrogen atoms in the structure were refined anisotropically. The H atoms on the C and N atoms were arranged geometrically.Table 1 summarizes the crystallology information of 1 and its refinement results.
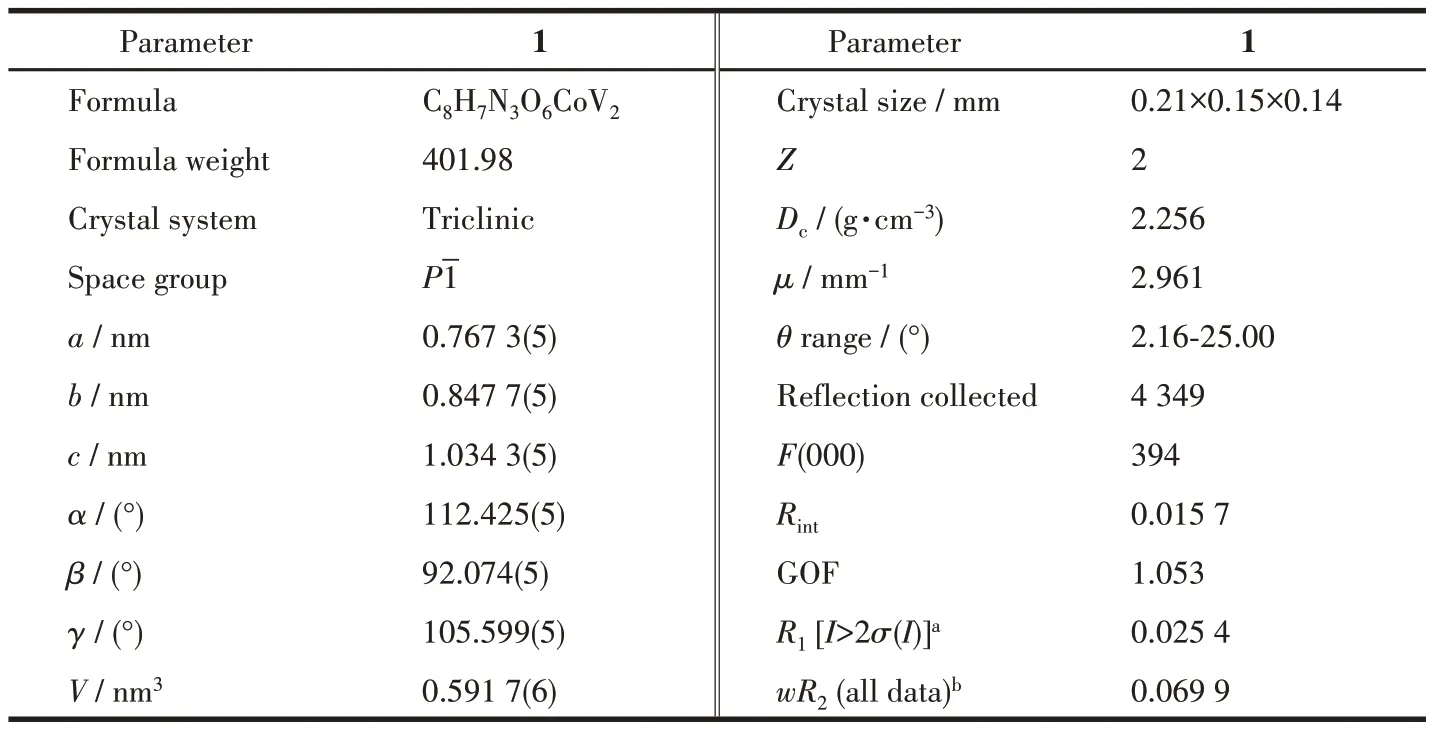
Table 1 Crystallographic data of 1 and corresponding structural refinements
CCDC:2285966.
2 Results and discussion
2.1 Synthesis and structure
X-ray single-crystal diffraction reveals the crystallization of 1 in the triclinicPspace group. The structure contains a crystallographically independent Co2+,a[V2O6]2-unit, and a pIM ligand. In this structure, Co2+coordinates with three O atoms and two N atoms from the ligand to form a twisted CoO3N2square pyramid configuration. There are two crystallographically different vanadium atoms:V1 and V2,both vanadiums adopt a distorted tetrahedral coordination pattern. Where, V1 coordinates with two bridging O atoms from two VO4,one bridging O from CoO3N2and the end O atoms of its own VO4, V2 coordinates with two bridged O atoms from CoO3N2and two bridged O atoms from VO4(Fig.1a). The average bond length of V—O is 0.172 9 nm, and those of Co—O and Co—N is 0.199 2 and 0.213 2 nm. The valence states of V and Co are determined to be +5 and +2 respectively through bondvalence sum calculations. An interesting feature of the structure is that the VO4tetrahedra and the CoO3N2tetragonal cone are connected by sharing O atoms to form a ternary ring system containing two fivemembered rings and one six-membered ring (Fig.1b).The five-membered ring includes four VO4tetrahedra and one CoO3N2tetragonal cone, while the sixmembered ring includes four VO4tetrahedra and two CoO3N2tetragonal cones. These ternary rings are further pointed and coplanar to form a 2D layer network(Fig.1c).
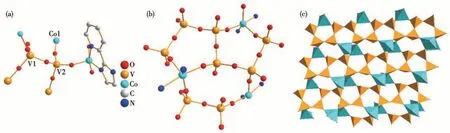
Fig.1 (a)Coordination for the Co and V in 1;(b)Ternary ring system containing two five-membered rings and one six-membered ring;(c)2D layer formed by ternary rings
2.2 IR spectra,PXRD and TG analysis
The IR spectra of 1 were studied in a range of 4 000-400 cm-1using a KBr disc (Fig.2a). The absorption peaks at 978 and 928 cm-1are attributed to the vibrations ofνas(V—O—V) and the absorption peaks at 841 and 646 cm-1are assigned to the vibrations ofνas(V—O—Co). The absorption peaks at 963, 882, and 835 cm-1belong to V=Ot(Oterminal) vibration, the region from 1 621 to 1 308 cm-1corresponds to the ligand C—C and C—N stretching vibration[28-30]. To further check the repeatability and purity of the crystal, the recovered crystalline samples were crushed as a fine powder for PXRD analysis. Compared with the crystal structure, the experimental PXRD patterns of the samples were in good agreement with the crystal simulation results, indicating that the bulk powders were pure phase (Fig.2b). The TG test of 1 was conducted in N2atmosphere at a heating of 10 ℃·min-1. The TG curve exhibited a sustained weight loss of 34.2% (calculated value 33.8%) between 335 ℃and 775 ℃, corresponding to the loss of pIM(Fig.3).
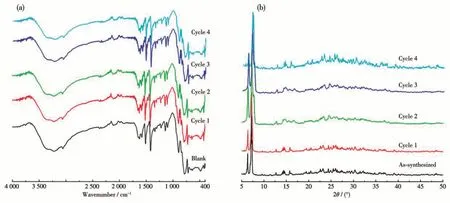
Fig.2 (a)IR spectra of 1 after each catalytic cycle;(b)PXRD patterns of recovered 1
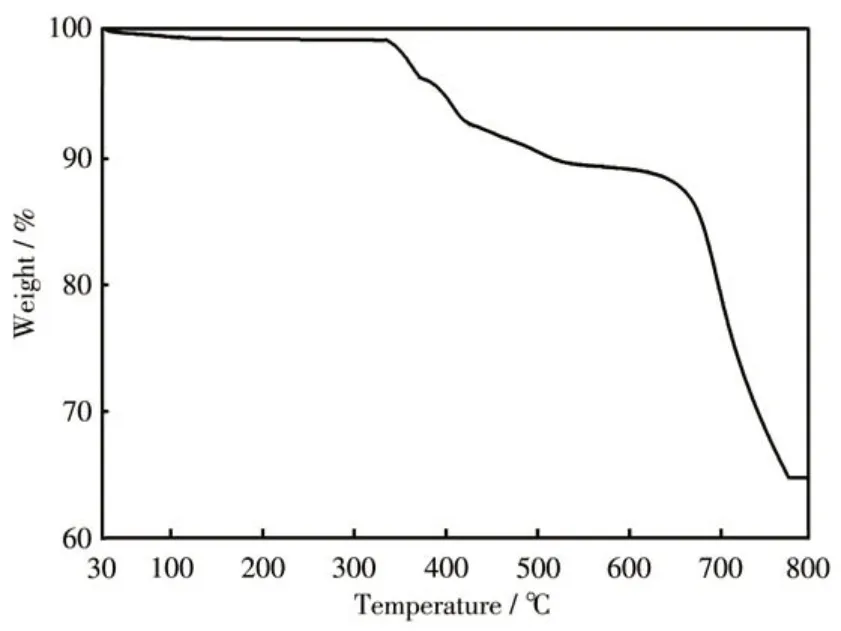
Fig.3 TG curve of 1 measured from 30 to 800 ℃under N2 atmosphere
2.3 Epoxidation of olefins
The catalytic epoxidation of olefin is affected by many factors, such as the dosage of the catalyst, temperature, reaction time, and the amount of oxidant.Therefore, we need to find the best reaction conditions to improve the conversion and selectivity of the product. Under gentle conditions, the olefins were oxidized in acetonitrile (CH3CN) with 1 as a heterogeneous catalyst and H2O2as an oxidant. An initiatory study on the oxidation of the cyclooctene to cyclooctane epoxide was selected to explore the catalytic activity of 1 in CH3CN at 60 ℃.Under the above conditions,the dosage of oxidant and catalyst was determined through controlled experiments. As shown in Fig.4a, the conversion of the epoxidation increased from the beginning 84.8% (0.01 mmol catalyst) to 98.6% (0.04 mmol catalyst). When the dosage of the catalyst added up to 0.07 mmol, the conversion remained nearly constant, suggesting that the appropriate dosage of the catalyst was only 0.04 mmol. Then, we examined the conversion for different dosages of oxidant. The conversion increased from 77.2% to 98.6% with the increase in the amount of oxidant from 0.5 mmol to 1.5 mmol (Fig.4b), however, the conversion did not improve significantly with further increase of H2O2dosage. According to the above test results,the most reasonable conditions for catalytic oxidation of cyclooctene are available 0.04 mmol catalyst and 1.5 mmol oxidant. So, we got optimum reaction conditions using 1 as the catalyst(0.04 mmol)and 30%H2O2as the oxidant (1.5 mmol) in CH3CN at 60 ℃(Scheme 1). As reflected in Table 2, 1 could availably catalyze cyclooctene to cyclooctane epoxide with the conversion of 98.6% and selectivity of 99.2% after 8 h of reaction, which was comparable to the previously reported POVs-based hybrids,such as[Zn(pIM)3]2V4O12·H2O,[Zn(ipIM)3]2V4O12,and[Co(eIM)3]2V4O12·H2O[23].
Scheme 1 Epoxidation of cyclooctene catalyzed by 1
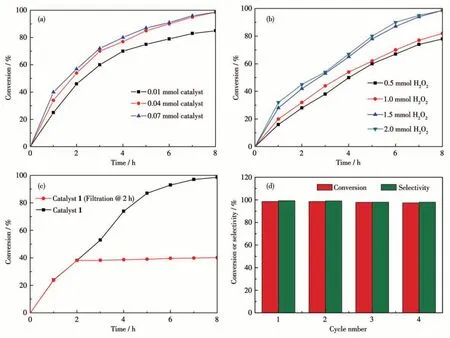
Fig.4 Effect of(a)catalyst and(b)oxidant factors for cyclooctene oxidation;(c)Thermal filtration experiment;(d)Recycling of catalyst for oxidation of cyclooctene
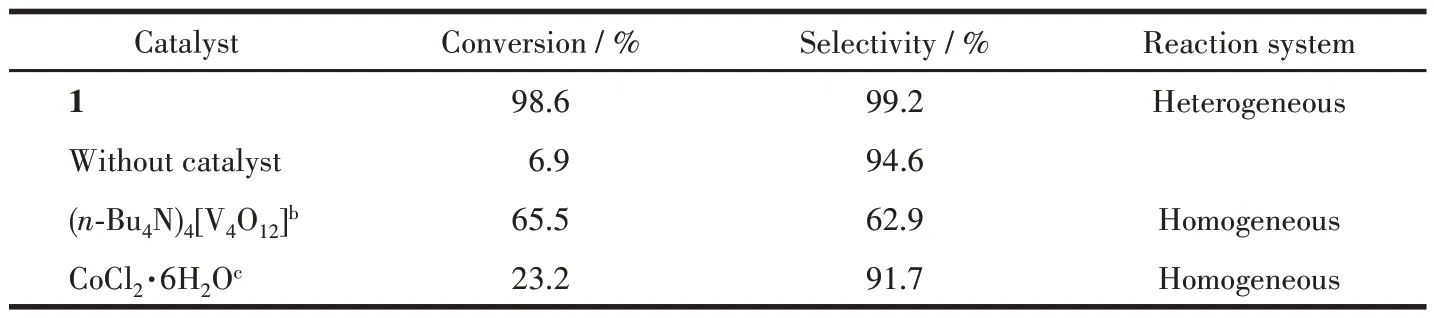
Table 2 Epoxidation of cyclooctene with different catalystsa
To further explore the role of the Co-complex and vanadium-oxygen anion in the catalytic reaction, contrast experiments were carried out, and the CoCl2·6H2O, (n-Bu4N)4[V4O12] were also used as a catalyst to explore catalytic activity. When CoCl2·6H2O was used as a catalyst, the cyclooctene conversion was very low,while(n-Bu4N)4[V4O12] produced a different result: the cyclooctene conversion achieved 65.5% (Table 2).From the above results, we could conclude that the combination of Co2+and V—O cluster by the complexation may cause a positive synergistic catalysis and significantly increase catalytic activity, this was similar to the previous report[31]. Besides, the catalytic activity might be also related to the unsaturated coordination sites of the Co2+, which could interact with the substrate to facilitate chemical reactions. Again, when the reaction was carried out without catalyst, only 6.9%conversion was observed,which indicated that the catalyst was vital for the reaction. According to the above catalytic results and literature reports[32-33], a possible epoxidation mechanism was suggested using 1 as a catalyst(Scheme 2).Primary,the coordinatively unsaturated Co2+in the structure as Lewis acidic centers available activated the olefin substrate, which not only pi-electron delocalization to the metal center but also shortened the distance between substrate and the peroxovanadium groups, the four-coordinated V5+simultaneously reacted with H2O2to generate active peroxovanadium groups, then, the O atom in peroxovanadium nucleophilic attacked the olefin double bond forming the epoxidation products and the catalytic cycle completed.

Scheme 2 Proposed mechanism of catalytic epoxidation procedure
To support the heterogeneous nature of the catalyst, a hot filtering test was conducted during the cyclooctene epoxidation. The solid catalyst 1 was separated from the reaction system after 2 h of reaction,and the filter was kept reacting for another 6 h with this understanding. The obtained filter was monitored by gas chromatography (GC) analysis, and the conversion was almost immobile (38.2%), which was significantly lower than the value in the presence of 1 (Fig.4c). The result confirmed the heterogeneous nature of the reaction system. Due to the excellent catalytic properties, 1 was chosen to test the cycling stability in heterogeneous systems. After the reaction was completed, the catalyst could be recovered easily from the reaction system through filtering and further reused in the subsequent epoxidation reaction. 1 could be recirculated at least four times without significant reduction in activity(Fig.4d). The combination of IR and PXRD patterns(Fig.2)before and after catalysis certified that the structure and crystallization remained unaltered after the circular reactions, which indicated excellent cycling stability of the catalysts.
Subsequently, various olefins were selected to estimate the catalytic universality of the catalyst. As shown in Table 3, the catalytic action of cyclohexene was examined under the same conditions, the slightly lower conversion achieved 94.2% within 6 h (entry 2),and the epoxidation yield was also slightly lower than that of cyclooctene (entry 1). So, the catalyst exerted excellent activity on cycloolefins.
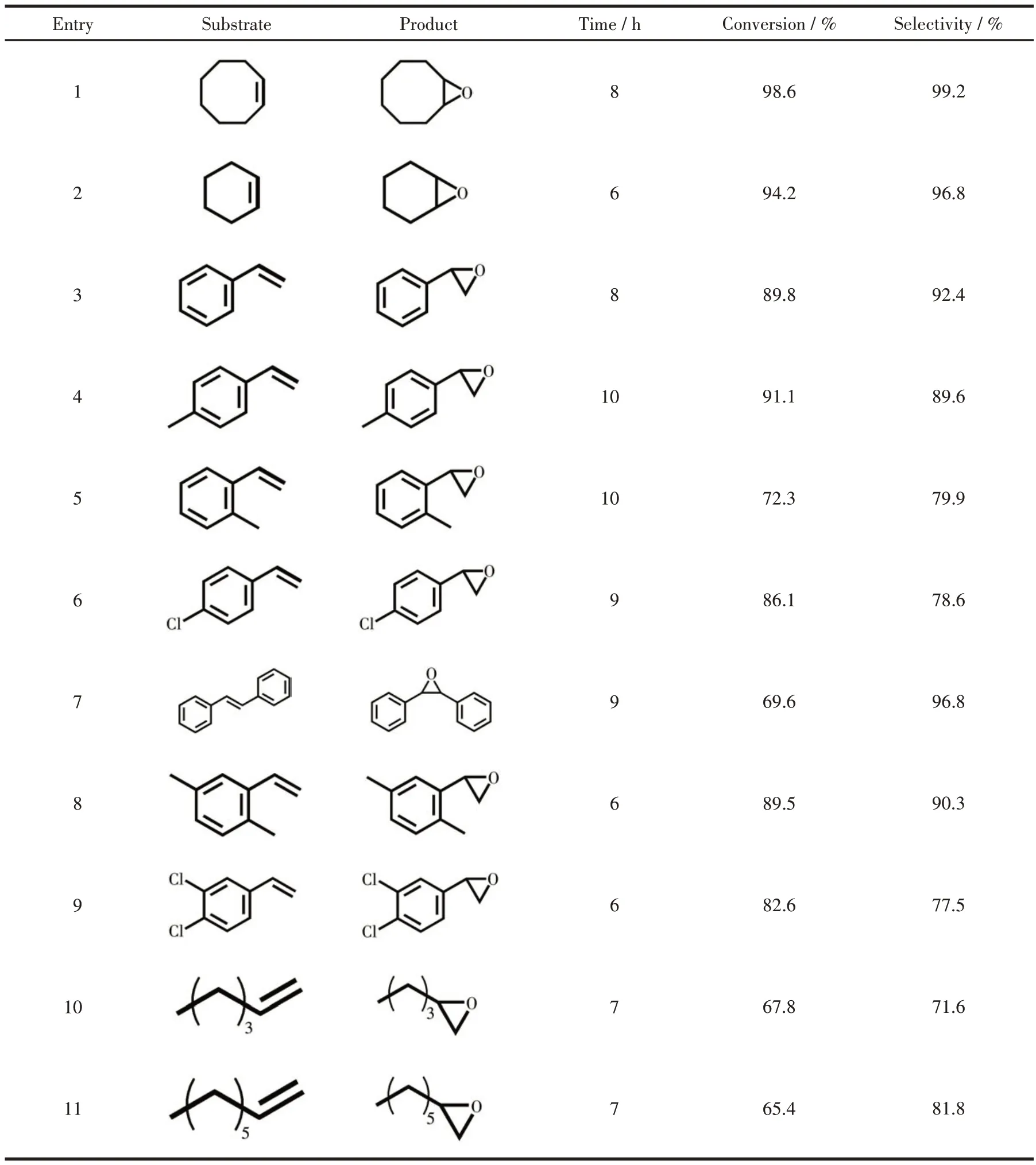
Table 3 Oxidation of various olefins catalyzed by 1 using H2O2 oxidant
However,for aromatic olefins,the effect of the catalyst was lower than that for cycloolefins. Under optimal contexts, the conversion of styrene was 89.8% with a narrowly satisfying selectivity of 92.4% for 8 h (entry 3). Middling catalytic activities for the oxidation ofp-methylstyrene (conversion 91.1%, selectivity 89.6%)ando-methylstyrene (conversion 72.3%, selectivity 79.9%) (entries 4-5) were given after 10 h of catalytic reactions. Compared with styrene andp-methylstyrene,a relatively lower catalytic was observed for the electrondeficientp-chlorostyrene(conversion 86.1%,selectivity 78.6%)(entry 6)for 9 h.The catalytic activity of multi-substituted and large steric hindrance substances was also studied,trans-stilbene afforded obvious reduced activity with 69.6% conversion for 9 h, probably due to a larger steric resistance containing diphenyl groups(entry 7).The reaction of 2,5-dimethylstyrene exhibited 89.5% conversion and 90.3% selectivity for 6 h (entry 8),compared with 2,5-dimethylstyrene,3,4-dichlorostyrene resulted in a relatively lower reduced activity with 82.6% conversion and 77.5% selectivity at the same time (entry 9). As for aliphatic linear olefins, the 1-hexene was transformed into the corresponding epoxide with 67.8% conversion and 71.6% selectivity for 7 h, and the reaction of 1-octene afforded 65.4% conversion and 81.8% selectivity for 7 h (entries 10-11). The above results show that the nature of the substrates is an important element affecting the epoxidation, and the catalytic oxidation of the circular substrates is more effective than that of the aromatic and linear substrates during the epoxidation process[34].
2.4 Magnetic measurements
The variable temperature magnetic susceptibility(χM) of 1 was conducted with a field intensity of 1 kOe.Fig.5a showed theχMTplot againstTat temperatures variable between 2-300 K.WhenTwas 300 K,theχMTvalue was 2.27 emu·K·mol-1, which was slightly higher than the theoretical spin-only value of 1.875 emu·K·mol-1for the high-spind7Co2+ions (S=3/2,g=2.0),implying the existence of an orbital angular momentum contribution[35-37]. Gradually lowered the temperature,theχMTvalue decreased gently to 2.12 emu·K·mol-1at 45 K, the curvilinear relationship betweenχMTandTimplied the intramolecular antiferromagnetic coupling among the Co2+centers.Then theχMTvalue dramatically descended to a minimum of 0.66 emu·K·mol-1at 2 K,this might be attributed to the integrated action of the magnetic anisotropy and spin-orbit coupling of Co2+as well as the antiferromagnetic interactions[38-40]. As displayed in Fig.5b, the linear fitting of theχM-1vsTkept to the Curie-Weiss law between 300-10 K, with the Curie constantCof 2.26 emu·K·mol-1and the Weiss constantθof -2.22 K, farther notarizing that the antiferromagnetic effect present in 1.

Fig.5 (a)Temperature reliance of χM and χMT for 1;(b)Temperature reliance of χM-1 for 1
3 Conclusions
A cobalt-vanadates architecture was hydrothermally prepared, containing cobalt nodes and V—O sheets. The compound was used as the catalyst for the olefins epoxidation and hydrogen peroxide was used as the oxidant. The catalytic results show that the compound has excellent epoxidation catalytic performance under optimized conditions, and can be recycled many times. The studies of other potential catalytic reactions using the compound were ongoing. Besides, magnetic measurements reveal the antiferromagnetical interactions between the Co2+ions.
Acknowledgments:This work was supported by the Basic Research Project Fund of Shanxi Province (Grant No.202203021222296),the Scientific and Technological Innovation Programs of Higher Education Institutions in Shanxi (Grants No. 2023L254, 2022L425), the Foundation of Shanxi Datong University (Grants No.2017-B-04, 2019-B-11, 2022Q24), the Key Research and Development Project of Datong (Grant No.2023003).
