从“湿热致瘀”角度探讨幽门螺旋杆菌感染对慢性萎缩性胃炎Hedgehog及NOX/NF-κB/STAT1信号通路的影响*
2024-04-16徐晓惠闫海琳徐子萱周姝含吕文亮
徐晓惠,闫海琳,徐子萱,周姝含**,吕文亮**
(1.桐乡市第一人民医院 桐乡 314500;2.湖北中医药大学中医临床学院 武汉 430061)
胃癌可分为两种亚型,较多见的是由萎缩性胃炎和肠上皮化生演变而来即遵循Correa发展模式的肠型胃癌。幽门螺旋杆菌(Helicobacter pylori,Hp)感染是我国胃癌的最主要原因,存在Hp 感染的萎缩性胃炎(Chronic atrophic gastritis,CAG)患者是肠型胃癌的高危人群,Hp 阳性的轻/中度萎缩和重度萎缩患者比Hp阴性患者的胃癌发病率高6.4 倍和11.8 倍[1]。由于炎症可控,而癌症难控,对包括CAG 在内的Hp相关性胃癌前病变的早期管理成了胃癌防控的着力点,对Hp相关性CAG的“炎癌转化”机制研究显得尤为重要。
目前,Hp 相关性CAG 的“炎癌转化”机制的相关研究中,在哺乳动物体内刺猬蛋白-细胞表面受体Ptch-G 蛋白偶联受体样蛋白Smo-胶质瘤相关癌基因同源蛋白Gli(Hh-Ptch-Smo-Gli)构成的信号通路(Hedgehog信号通路)异常活化被认为与CAG 的“炎癌转化”有关[2]。胶质瘤相关癌基因同源蛋白1(Gli1)在核内促发Shh下游靶基因和自身Gli基因的转录,诱导DNA 的异常复制而导致肿瘤发生[3],在包括胃癌在内的多种癌症中,均出现Hh-Ptch-Smo-Gli 通路的过度激活[4]。
此外,炎症因子的释放与“炎癌转化”密切相关,NADPH 氧化酶(NOX)作用是在免疫防御中产生活性氧(Reactive oxygen species,ROS),抑制病原体入侵[5],肿瘤细胞发育过程中会产生过量的ROS[6],促进肿瘤新生血管的生成,诱发或启动包括炎症和肿瘤在内的多种疾病[7-9]。NF-κB 信号通路与机体炎症因子的释放,肿瘤的发生、生长和转移等多个过程密切相关[10]。转录信号转导子和转录活化子1(Signal transducer and activator of transcription 1,STAT1)被认为是一个抑癌因子,可促肿瘤细胞凋亡[11],并可调控NF-κB 转录因子的表达[12],且NF-κB 和STAT1 信号通路的激活受ROS信号调控[13]。NF-κB/STAT1信号通路在萎缩性胃炎“炎癌转化”过程中可能发挥着不可忽视的作用。
从中医辨证的角度而言,Hp被认为是一种湿热性质的病邪,Hp 感染与脾胃湿热证关系最密切,因为饮食失节等使脾胃受损,湿热内生,更利于Hp 的定植,感染人体后造成黏膜损伤又会进一步加重“湿热证”,课题组前期进行了慢性胃炎的病机转化规律研究,发现脾胃湿热-胃络瘀血的病机转化位于慢性萎缩性胃炎腺体萎缩-肠化生的关键节点。湿热不去而酿毒损络成瘀,胃黏膜病变渐生,瘀血与湿热蕴结,久化瘀毒,瘀血证的出现常被认为是黏膜恶变的重要标志。
因此,本研究着眼于Hp 相关性CAG 和非Hp 感染的CAG 患者,运用蛋白质免疫印迹(Western blot)和实时荧光定量PCR(RT-qPCR)检测其胃黏膜中Hh-Ptch-Smo-Gli信号通路与烟酰胺腺嘌呤二核苷酸磷酸氧化酶/核因子κB/转录信号转导子和转录活化子1(NOX/NF-κB/STAT1)通路的关键因子,从湿热致瘀角度探究Hp感染后CAG“炎癌转化”的作用机制。
1 研究对象与方法
1.1 患者来源及分组
患者来源于2021 年8 月-2021 年12 月湖北中医药大学附属湖北省中医院脾胃病科CAG 患者,分为CAG 伴Hp 感染组(HP+CAG 组,n=21)与CAG 不伴Hp感染组(HP-CAG组,n=22)。
1.2 诊断标准
参考《慢性萎缩性胃炎中西医结合诊疗共识意见(2017 年)》[14]、《第五次全国幽门螺杆菌感染处理共识报告》[15]、《成人幽门螺杆菌引起的胃炎中西医协作诊疗专家共识(2020,北京)》[16],根据临床症状、胃镜、病理活检、尿素呼气试验、快速尿素酶试验或Hp 培养等结果诊断。
1.3 伦理委员会批准
本研究经湖北省中医院伦理委员会批准(HBZY2021-C15-01)。参与者均详细了解此项研究内容及相关权利后签署知情同意书。
1.4 胃黏膜样本采集
胃镜下使用一次性活检钳取约1 mm×1 mm×1 mm的距幽门2-3 cm 胃窦临近胃小弯一侧的黏膜组织3 块,其中2 块分别放入无菌的冻存管内,迅速投入液氮中冻存,后存放于-80℃冰箱保存备用,另1 块使用4%多聚甲醛以固定组织,常温保存,后以石蜡包埋、切片备用。
1.5 RT-qPCR引物序列
具体引物序列见表1。GAPDH 表达做内部参照,使用PCR 仪分析cDNA,并绘制溶解曲线,以QPCR 算法(相对定量,2-ΔΔCt法)进行数据分析。QPCR 算法公式:

表1 引物序列表
1.6 数据分析
实验数据采用SPSS 25.0软件进行统计分析,计数资料采用百分比表示,计量资料以平均值±标准差(±s)表示。两组间的差异比较,若方差齐,采用T检验(Student'st-test);若方差不齐,采用Wilcox 秩和检验(Wilcoxon rank-sum test),多组间均数行单因素方差分析(One-way ANOVA),以P<0.05 为差异具有统计学意义。
2 实验结果
2.1 研究对象分组情况基本资料
Hp+CAG 与Hp-CAG 组患者性别比例与年龄无统学差异(P>0.05),具体可见表2。
表2 研究对象分组情况基本资料(±s)

表2 研究对象分组情况基本资料(±s)
组别Hp+CAG组(n=21)Hp-CAG组(n=22)性别男(n)11(52.4%)10(45.5%)女(n)10(47.6%)12(54.5%)年龄(岁)54.33±6.79 51.89±7.24
2.2 两组患者胃黏膜病理表现
胃镜下观察两组患者胃黏膜红白相间、以白为主,粘膜变薄或粗糙不平,其中可见血管纹理,或伴有点、片状红斑或散在出血点,部分患者可见黏膜水肿。
苏木精-伊红(HE)染色后,在镜下观察各组患者黏膜组织病理情况,结果显示:Hp+CAG组患者胃黏膜出现上皮细胞部分脱落,腺体数量减少,排列紊乱,可见明显肠上皮化生及炎症细胞浸润;Hp-CAG 组患者胃黏膜腺体数量减少,排列紊乱,局部可见肠上皮化生,炎症细胞浸润程度较Hp+CAG组轻(见图1)。
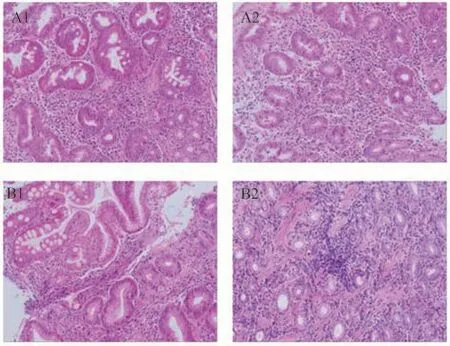
图1 各组患者胃黏膜组织HE染色(×200)

图2 两组蛋白相对表达量
参照慢性胃炎新悉尼系统对胃黏膜炎症程度和活动度进行分级(见表3,表4),统计两组患者胃黏膜出现不同程度和活动度炎症的例数并进行统计学分析。结果表明,Hp+CAG 组患者胃黏膜均出现不同程度的炎症,且出现重度炎症的比例明显高于Hp-CAG组(P<0.05);轻度或无炎症患者的比例低于Hp-CAG组(P<0.05);两组患者中胃黏膜出现中度炎症的比例相当,差异没有统计学意义(P>0.05)。Hp+CAG 组患者胃黏膜出现重度和中度炎症活动度的比例明显高于Hp-CAG 组(P<0.05);而出现轻度或无炎症活动度的比例明显低于Hp-CAG组(P<0.05)。

表3 两组患者胃黏膜炎症程度分级
2.3 RT-qPCR检测结果
经数据分析发现,两组患者胃黏膜Gli1 mRNA、Gli2 mRNA、Gli3 mRNA 水平有显著差异(P<0.01)(见表5)。两组患者胃黏膜Shh mRNA、Smo mRNA、Ptch mRNA差异具有统计学意义(P<0.01)(见表6)。两组患者胃黏膜NOX1 mRNA、NOX2 mRNA、NOX4 mRNA 与NFκB mRNA差异具有统计学意义(P<0.01)(见表7)。
表5 两组患者胃组织Gli1、Gli2、Gli3 mRNA检测结果(±s)

表5 两组患者胃组织Gli1、Gli2、Gli3 mRNA检测结果(±s)
注:与Hp+CAG组比较,**P<0.01。
Gli3 1.056±0.050 0.438±0.008**组别Hp+CAG组(n=21)Hp-CAG组(n=22)Gli1 1.046±0.040 2.700±0.017**Gli2 0.985±0.048 0.463±0.011**
表6 两组胃组织Shh、Smo、Ptch mRNA检测结果(±s)

表6 两组胃组织Shh、Smo、Ptch mRNA检测结果(±s)
注:与Hp+CAG组比较,**P<0.01。
Ptch 1.052±0.045 2.719±0.026**组别Hp+CAG组(n=21)Hp-CAG组(n=22)Shh 1.089±0.077 2.863±0.025**Smo 1.050±0.043 2.717±0.023**
表7 两组胃组织NOX1、NOX2、NOX4、NF-κB mRNA检测结果(±s)
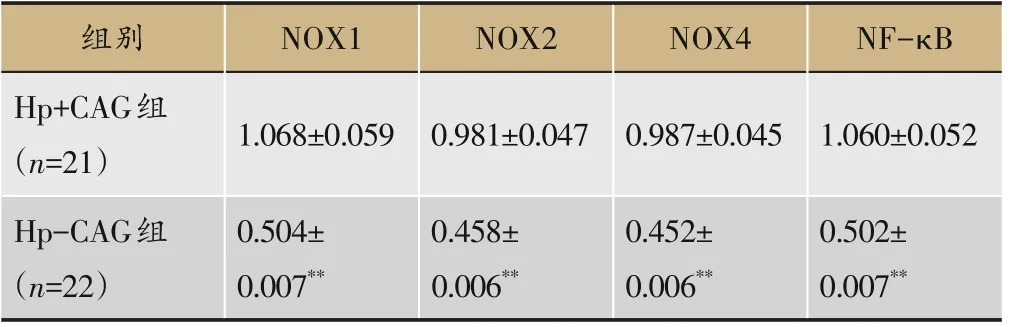
表7 两组胃组织NOX1、NOX2、NOX4、NF-κB mRNA检测结果(±s)
注:与Hp+CAG组比较,**P<0.01。
组别Hp+CAG组(n=21)Hp-CAG组(n=22)NOX1 NOX2 NOX4 1.068±0.059 NF-κB 0.981±0.047 0.987±0.045 1.060±0.052 0.502±0.007**0.504±0.007**0.458±0.006**0.452±0.006**
2.4 Western blot检测结果
两组患者胃黏膜NOX1/GAPDH、NOX2/GAPDH、NOX4/GAPDH 相对表达量差异具有统计学意义(P<0.01)(见表8)。两组患者胃黏膜p-P65/GAPDH 相对表达量、STAT1/GAPDH 相对表达量差异具有统计学意义(P<0.01);两组P65/GAPDH 相对表达量的差异没有统计学意义(P>0.05)(见表9)。
表8 两组NOX1/GAPDH、NOX2/GAPDH、NOX4/GAPDH相对表达量(±s)

表8 两组NOX1/GAPDH、NOX2/GAPDH、NOX4/GAPDH相对表达量(±s)
注:与Hp+CAG组比较,**P<0.01。
NOX4/GAPDH 0.800±0.054 0.474±0.094**组别Hp+CAG组(n=21)Hp-CAG组(n=22)NOX1/GAPDH 0.757±0.139 0.393±0.114**NOX2/GAPDH 0.561±0.073 0.231±0.029**
表9 两组STAT1/GAPDH、P65/GAPDH、p-P65/GAPDH相对表达量(±s)

表9 两组STAT1/GAPDH、P65/GAPDH、p-P65/GAPDH相对表达量(±s)
注:与Hp+CAG组比较,**P<0.01。
p-P65/GAPDH 0.599±0.163 0.265±0.149**组别Hp+CAG组(n=21)Hp-CAG组(n=22)STAT1/GAPDH 0.134±0.091 0.466±0.115**P65/GAPDH 0.851±0.055 0.780±0.078
3 讨论与结论
本次研究重点观察了幽门螺旋杆菌感染对慢性萎缩性胃炎患者胃黏膜炎症程度的影响,显而易见,Hp 感染明显加重了CAG 患者胃黏膜炎症程度,与此同时,Hp 感染后NOX/NF-κB 信号通路的异常激活及Hh-Ptch-Smo-Gli 信号通路的抑制,使“炎癌转化”风险增加。
Hp+CAG组患者胃黏膜出现中度及重度炎症的比例明显高于Hp-CAG 组,提示Hp 感染明显加重了CAG 患者胃黏膜炎症程度。有报道称轻度、中度及重度胃黏膜炎症患者的Hp 感染率逐渐升高[17]。Correa's Cascade 相关胃黏膜病变(包括萎缩性胃炎、肠上皮化生、不典型增生和胃癌)中Hp 感染率均高于非萎缩性胃炎组(P<0.01),且胃黏膜病变程度随着Hp感染程度分级的增加而增加[18],说明胃黏膜病变程度与Hp感染程度有关。
Hp通过释放尿素酶来中和胃内的酸性环境,在胃中长期存活[19],同时利用鞭毛和趋化受体在胃黏膜上皮细胞定植,导致炎症和中性粒细胞浸润[20-21]。此外,Hp凭借其毒力因子细胞毒素相关蛋白A(CagA)、空泡毒素A(VacA)等,干扰宿主细胞内信号通路,导致胃黏膜长期慢性炎症,参与CAG 发生发展,驱动“炎癌转化”[22-23]。例如,CagA 在感染的细胞中激活NF-κB 依赖的炎症信号传导,促进白细胞介素-8(IL-8)的产生[24-25],导致中性粒细胞和巨噬细胞的招募,聚集的中性粒细胞、单核巨噬细胞会产生大量的NOX2,继而生成过量的ROS[26]。由于Hp 菌体内含有各种超氧化物还原酶,具有较强的抗氧化能力,更关键的是,Hp会破坏NOX 的靶向性[27],使超氧阴离子释放到细胞外环境中,而不会在Hp 吞噬体内积累,所以Hp 感染导致的NOX2 异常活化,只会加速胃黏膜的氧化损伤而不影响Hp 的继续定植。故宿主对其的氧化杀伤往往失败,结果Hp 感染持续存在,而ROS 生成和释放持续增多,导致胃黏膜上皮细胞持续损伤[28]。
NOX 的过度激活是导致氧化应激水平升高的重要机制,其中NOX2 在人胃黏膜的表达与胃黏膜的炎症程度和萎缩损伤程度呈正相关[29]。有研究发现NOX4 通过产生ROS 和激活Gli1信号在胃癌细胞生长和凋亡中起重要作用[12]。NOX1、NOX2 与NOX4 也被认为参与了血管生成的各个阶段,在癌症诱导的血管形成中发挥着关键作用[13]。NF-κB是调控基因转录的重要因子,p65蛋白是NF-κB中的一种重要转录因子,被认为广泛参与了细胞生长、分化、炎症、免疫应答等多方面的表达调控,其过度表达与多种慢性炎症性疾病及肿瘤发病密切相关,磷酸化的p65蛋白(p-P65)是p65 蛋白的活化形式。有报道称Hp 感染后NF-κB 呈激活状态,且萎缩性胃炎患者胃黏膜萎缩病变程度与Hp 感染和p-P65 的表达呈正相关[30],随着胃黏膜病变程度的加重,胃黏膜中p-P65 表达及Hp 阳性率均增高[31]。活化NF-κB 的因素有很多,包含病原体、ROS等,其中NOX是一个关键的酶系统,NOX2等过表达会产生过量的ROS 激活NF-κB 信号通路,同时促进STAT 磷酸化。STAT1 已被证明通过调节免疫系统和促进肿瘤免疫监测来发挥抗肿瘤作用,是肿瘤血管发生、生长和转移的负调节因子,被认为是潜在的肿瘤抑制因子,现多认为其可促进肿瘤细胞凋亡,抑制胃癌细胞增殖及侵袭、迁移能力[32-33],有研究显示Hp 感染后明显抑制STAT1 核易位[34];且胃癌患者血清STAT1 表达水平显著低于慢性胃炎组和健康对照组[35]。
如今人们居住条件和生活水平大大提高,过食肥甘厚味,脑力劳动偏多,脾气不健,加之作息不规律,湿热内蕴常见。作为慢性萎缩性胃炎的重要致病因素,Hp 也被认为是一种湿热性质的伏邪,正气不甚虚时,其隐匿于胃部伏而不发,长期感染造成胃黏膜的持续炎症,加重黏膜病理改变[36]。
湿邪日久伤阳,热邪日久耗阴,湿热胶结难化,缠绵体内导致阴阳俱损,中气更虚,气血不荣则黏膜萎缩、纳运失司,湿热不去而酿毒损络成瘀,胃黏膜病变渐生,瘀血与湿浊、湿热蕴结,久化瘀毒,裹挟留滞[37],瘀血证的出现常被认为是黏膜恶变的重要标志。
Hp+CAG 组患者胃黏膜NOX1 mRNA、NOX2 mRNA、NOX4 mRNA、NF-κB mRNA 与p-P65 水平显著增高,STAT1 表达水平降低。说明Hp 感染后NOX/NF-κB 信号通路异常激活,促进组织损伤与促炎因子的释放,使萎缩性胃炎患者胃黏膜炎症程度和活动度增加,而STAT1 水平显著降低,“炎癌转化”风险增加。此外,Hp 感染使CAG 患者胃黏膜Gli1 mRNA、Shh mRNA、Smo mRNA、Ptch mRNA 水平显著降低,同时Gli2 mRNA 与Gli3 mRNA水平显著增高,说明Hp感染后CAG 患者胃黏膜Hh-Ptch-Smo-Gli 信号通路可能受抑制。有研究表明Hp 感染后可能通过下调胃黏膜Shh 蛋白的表达导致胃黏膜萎缩及肠上皮化生(IM)[38]。Shh在CAG 肠化生组织中表达率低于在非萎缩性胃炎胃组织中的表达率[39-40],也提示Shh分泌不足与胃黏膜肠上皮化生有关。
在CAG“炎癌转化”过程中,不可忽视的环节是源自骨髓的间充质干细胞(MSCs)被招募到组织损伤部位。Donnelly 等[41]发现MSCs 在Hp 感染的小鼠胃发育不良腺体中增殖,出现肠化生和异型增生。当募集的MSCs 重新填充胃上皮时,炎症细胞因子持续存在而缺乏足够的Shh,导致胃上皮异常再生,出现异型增生和癌症[42]。Hp感染诱导的慢性炎症环境与Shh分泌不足或传导抑制是在CAG 发生发展过程中的两个关键因素。胃黏膜存在长期慢性炎症的情况下,Shh 信号通路的失调会进一步导致胃上皮分化异常和胃癌。不少研究证实,益气活血法可激活hedgehog 信号通路,对萎缩性胃炎癌前病变大鼠胃黏膜病变起到改善作用[43-44]。
总结而言,存在Hp 感染的萎缩性胃炎患者胃黏膜病变机率高,可能与胃黏膜长期炎症,以及Hp 感染下调Shh、Gli1、Smo mRNA 表达水平,上调Gli3 表达水平,抑制Hh-Ptch-Smo-Gli 信号通路,同时使NOX/NF-κB/STAT1信号通路异常活化有关。
