腺苷A2B受体激活减轻脓毒症诱导的急性肺损伤及肺微血管内皮炎症损伤
2024-04-13王慧霞安友仲
王慧霞, 安友仲
急性呼吸窘迫综合征(acute respiratory distress syndrome,ARDS)是由肺炎、非肺部感染、创伤、输血、烧伤、误吸或休克等危险因素诱发,导致肺血管内皮和上皮通透性增加而引起的急性、弥漫性、炎症性肺损伤[1]。脓毒症是并发ARDS病因之一,同时若脓毒症患者继发ARDS可导致病死率显著增高[2]。目前,脓毒症并发ARDS发生机制尚未明晰,且具有高度异质性,其中肺微血管内皮细胞炎症损伤及内皮屏障通透性改变是其病理生理机制中的重要环节[3-4]。腺苷受体(adenosine receptor,AR),包括A1、A2A、A2B和A3受体,以组织依赖性方式在多种组织和细胞中表达,在免疫系统功能障碍、情感障碍及炎症等方面具有潜在药理学作用[5-6]。肺内最主要的AR是腺苷A2B受体(adenosine A2Breceptor,A2BR),在微血管内皮细胞中高表达[7],亲和力低,在缺血缺氧及炎症损伤时可被激活。但A2BR激活对急性肺损伤(acute lung injury,ALI)及肺微血管内皮损伤的影响仍有待进一步探讨。本研究建立盲肠结扎穿孔(cecal ligation and perforation,CLP)致脓毒症引发的ALI动物模型,在动物体内水平探索A2BR对ALI的影响。另通过TNF-α处理原代培养的人肺微血管内皮细胞(human pulmonary microvascular endothelial cells,HPMECs),在体外从细胞水平研究A2BR激活对ALI肺微血管内皮的影响及其可能的作用机制,为寻找新的潜在治疗靶点策略奠定理论基础。
1 材料与方法
1.1实验动物 选用无特定病原体(specific pathogen free,SPF)级雄性Sprague-Dawley(SD)大鼠24只(6~8周龄,体重200~220 g),购自北京维通利华实验动物技术有限公司。大鼠饲养环境温度设为(22±2)℃,自然光暗周期,实验期间自由饮食。
1.2细胞及主要试剂 原代培养HPMECs及内皮细胞培养基(endothelial cell medium,ECM)购自美国ScienCell公司;苏木精-伊红(hematoxylin and eosin,HE)染色液购自昆明云科生物技术有限公司;Transwell购自美国Corning公司;肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)、白细胞介素-6(interleukin-6,IL-6)、白细胞介素-1β(interleukin-1β,IL-1β)酶联免疫吸附试验(enzyme-linked immunosorbent assay,ELISA)检测试剂盒购自上海江莱生物科技有限公司;放射性免疫沉淀分析(radioimmunoprecipitation assay,RIPA)裂解液、二辛可定酸法(bicinchoninic acid,BCA)蛋白浓度测定试剂盒购自北京索莱宝科技有限公司;增强型化学发光(enhanced chemiluminescence,ECL)试剂盒购自上海碧云天生物科技有限公司;兔抗IL-1β多克隆抗体、小鼠抗人细胞间黏附分子-1(intercellular adhesion molecule-1,ICAM-1)抗体、兔抗人促血管生成素(angiopoietin,ANGPT)抗体、兔抗人血管内皮钙黏连蛋白(vascular endothelial-cadherin,VE-cadherin)抗体、小鼠抗-3-磷酸甘油醛脱氢酶(glyceraldehyde-3-phosphate dehydrogenase,GAPDH)单克隆抗体购自英国Abcam公司;辣根过氧化物酶(horseradish peroxidase,HRP)标记的山羊抗兔IgG、山羊抗小鼠IgG购自北京中杉金桥生物技术有限公司;Recombinant Human TNF-α购自美国PEPROTECH公司;A2BR靶向激动剂BAY60-6583、A2BR抑制剂PSB1115购自美国ApexBio公司。
1.3实验方法
1.3.1 动物分组及模型制备 将24只SPF级SD雄性大鼠按随机数字表法分为假手术组(sham组)、ALI模型组(ALI组)、A2BR激动剂BAY60-6583干预组(ALI+BAY组)和A2BR抑制剂PSB1115干预组(ALI+PSB组),每组6只。采用CLP法建立脓毒症致ALI大鼠模型[8]:大鼠麻醉后开腹,寻找盲肠末端,用无菌4号线进行结扎,14G针头贯穿盲肠,挤出粪便及肠内容物后回纳腹腔,关腹。sham组仅进行开腹和关腹操作。ALI+BAY组及ALI+PSB组在术前30 min分别腹腔内注射100 μL生理盐水+2 mg/kg BAY60-6583、100 μL生理盐水+10 mg/kg PSB1115,其余各组经腹腔给予等量生理盐水,24 h后过量麻醉处死。本实验中动物处置方法符合动物伦理学标准,并经北京大学人民医院医学伦理委员会审批(批号:2019PHC014)。
1.3.2 细胞培养及干预方法 HPMECs快速解冻后,使用添加了5%胎牛血清(fetal bovine serum,FBS)、1%内皮细胞生长添加剂、100 IU/mL青霉素、100 IU/mL链霉素的ECM培养,培养箱环境条件设置为95%空气、5% CO2、37 ℃,及时更换培养基以满足细胞生长需要,待细胞生长密度达80%时进行消化传代,细胞生长状态稳定后取3~5代细胞用于实验。药物干预前血清饥饿24 h,并予1 kU/L腺苷脱氨酶预处理,以去除内源性腺苷,除外内源性激动腺苷受体的作用。将细胞分为6组:对照组、TNF-α组、BAY组、PSB组、BAY+TNF-α组和PSB+TNF-α组。BAY+TNF-α组提前1 h加入1 μmol/L的BAY60-6583预处理,PSB+TNF-α组提前1 h加入1 μmol/L的PSB1115预处理,TNF-α组、BAY+TNF-α组、PSB+TNF-α组加入TNF-α 100 ng/mL刺激24 h,收集细胞进行后续检测。
1.3.3 肺组织HE染色 大鼠过量麻醉处死,开胸取右上肺,置于中性甲醛中固定,脱水,石蜡包埋切片后行HE染色。光镜下观察肺组织病理改变并采用Smith法进行评分。Smith法是对肺水肿、肺泡以及间质炎症、肺泡及间质出血、肺不张和透明膜形成分别进行0~4分半定量分析。无损伤为0分,病变范围<25%为1分,25%≤病变范围<50%为2分,50%≤病变范围<75%为3分,病变范围≥75%至满视野为4分,总肺损伤评分为上述各项之和,每只动物观察10个高倍镜视野,取其平均值。
1.3.4 肺湿/干质量比值(wet/dry mass ratio,W/D)计算 大鼠过量麻醉处死,开胸取右下肺进行称量,此时右下肺重量记为湿重。将各组右下肺放入95 ℃烤箱连续烘干48 h,然后取出称量,该重量为肺干重。
1.3.5 肺水清除率(alveolar fluid clearance,AFC)测定 大鼠过量麻醉处死,气管切开进行气管插管,结扎右主支气管,将气管、肺和心脏整体取出,左肺用于测定AFC。将含有5%白蛋白和0.15 mg/mL Evans Blue 37 ℃提前预热的1.5 mL生理盐水通过气管插管注入左肺,并给予1 mL氧气以保证灌注液到达肺脏,将左肺用保鲜膜包裹后置于37 ℃水浴环境,并维持气道压在8 cmH2O,持续给予100%氧气,1 h后吸取左肺肺泡内液体,并于分光光度计波长620 nm处测定Evans Blue标记的白蛋白浓度并计算AFC。
1.3.6 肺组织炎症因子测定 取肺组织匀浆上清液,按照ELISA试剂盒说明书步骤测定TNF-α、IL-6和IL-1β含量。
1.3.7 异硫氰酸荧光素(fluorescein isothiocyanate,FITC)-白蛋白(albumin)法检测HPMECs单层通透性
HPMECs以2×105cells/mL密度接种于Transwell(孔径3.0 μm,直径24 mm)上室,相差显微镜下观察细胞形态及融合程度。待细胞完全融合48 h后进行同步化,各组在上室加入相应药物,在上室加入FITC-albumin,下室加入等摩尔、无FITC标记的albumin。37 ℃、5% CO2培养箱避光孵育1 h后,分别从上室、下室提取0.3 mL和1.2 mL样品。荧光分光光度计检测样品荧光能量值(吸收波长493 nm,发射波长528 nm)。制作FITC-albumin标准曲线,得到上室、下室FITC-albumin浓度,并计算HPMECs通透系数。
1.3.8 Western blot法检测蛋白表达 收集处理后的HPMECs,分别加入预冷RIPA裂解液,冰上孵育15 min后收集裂解组织或细胞液体,4 ℃、12 000 r/min离心20 min,取上清液即为总蛋白裂解液。按照BCA检测试剂盒说明书进行蛋白浓度检测,经加温处理后将蛋白样本与蛋白上样缓冲液进行混合,使用十二烷基硫酸钠-聚丙烯酰胺凝胶进行电泳实验,通过湿转法将蛋白转移至聚偏二氟乙烯膜(polyvinylidene fluoride,PVDF)上,用含5%脱脂奶粉的TBST溶液在室温下封闭2 h后切膜,分别加入IL-1β、ICAM-1、ANGPT、VE-cadherin和GAPDH一抗,4 ℃孵育过夜。次日以TBST溶液洗膜3次,10 min/次,二抗使用HRP标记的山羊抗兔IgG或山羊抗鼠IgG溶液,室温孵育2 h,TBST再次洗膜3次,10 min/次,加入化学发光显色。扫描并通过Image J软件分析其灰度值,以目标条带与内参GAPDH条带的灰度比值作为目标蛋白相对表达量,实验独立重复3次。
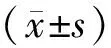
2 结果
2.1四组大鼠肺组织病理学改变及Smith评分比较
光镜下,sham组肺组织未见明显充血及出血,ALI组肺泡结构塌陷破坏,可见炎症细胞浸润和不同程度的肺间质、肺泡水肿、出血,见图1。ALI组Smith评分显著高于sham组(P<0.05)。ALI+BAY组Smith评分显著低于ALI组(P<0.05)。ALI+PSB组Smith评分显著高于ALI组(P<0.05)。见表1。
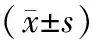
表1 四组大鼠Smith评分、肺W/D、AFC比较
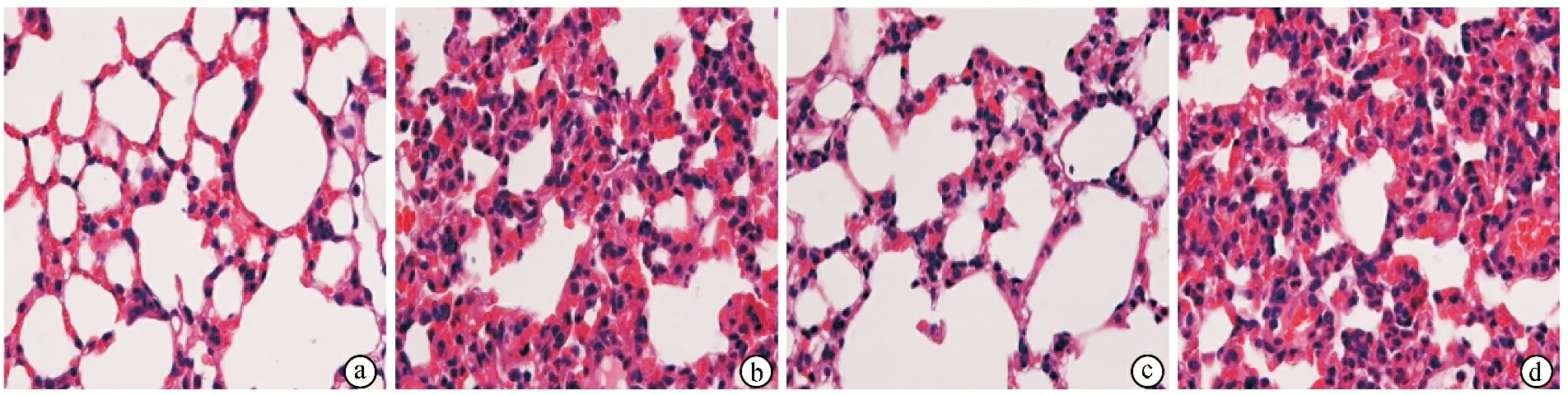
ⓐsham组肺泡结构完整且间隔正常,肺泡腔及间质未见明显炎症细胞渗出及水肿;ⓑALI组肺泡壁明显增厚伴大量中性粒细胞浸润,组织间隙可见出血,肺泡结构塌陷破坏,肺泡腔内可见炎症细胞浸润和液体潴留;ⓒALI+BAY组肺泡壁厚度明显减少,且中性粒细胞也随之减少,组织间隙可见少量出血,肺泡及肺间质少量炎症细胞浸润;ⓓALI+PSB组肺泡壁明显增厚伴大量中性粒细胞浸润,肺泡腔严重缩窄,组织间隙可见明显出血,一些肺泡内可见疑似透明膜形成
2.2四组大鼠肺W/D、AFC比较 与sham组比较,ALI组肺W/D显著升高,AFC显著降低(P<0.05)。与ALI组比较,ALI+BAY组肺W/D显著降低,AFC显著升高(P<0.05);ALI+PSB组肺W/D显著升高,AFC显著降低(P<0.05),见表1。
2.3四组大鼠肺组织炎症因子水平比较 与sham组比较,ALI组肺组织TNF-α、IL-6、IL-1β水平均显著升高(P<0.05)。与ALI组比较,ALI+BAY组肺组织TNF-α、IL-6、IL-1β水平均显著降低(P<0.05),而ALI+PSB组肺组织TNF-α、IL-6、IL-1β水平均显著升高(P<0.05),见图2。

与sham组比较,*P<0.05;与ALI组比较,#P<0.05
2.4各组HPMECs单层通透性比较 与对照组比较,TNF-α组HPMECs单层通透性显著增加(P<0.05)。与TNF-α组比较,BAY+TNF-α单层通透性显著降低,PSB+TNF-α组单层通透性显著增加(P<0.05)。BAY60-6583和PSB1115本身对HPMECs通透性无显著影响。见图3。
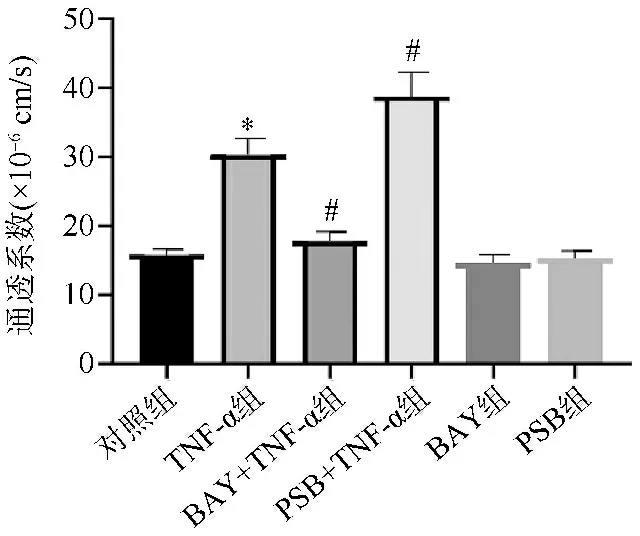
与对照组比较,*P<0.05;与TNF-α组比较,#P<0.05
2.5各组HPMECs的IL-1β、ICAM-1、ANGPT以及VE-cadherin表达水平比较 与对照组比较,TNF-α组IL-1β、ICAM-1表达水平显著升高(P<0.05)。与TNF-α组比较,BAY+TNF-α组IL-1β、ICAM-1表达水平显著降低,而PSB+TNF-α组IL-1β、ICAM-1表达水平显著升高(P<0.05)。BAY60-6583和PSB1115本身对IL-1β、ICAM-1表达无显著影响。与对照组比较,TNF-α组ANGPT、VE-cadherin表达水平显著降低(P<0.01),与TNF-α组比较,BAY+TNF-α组ANGPT、VE-cadherin表达水平显著升高,而PSB+TNF-α组ANGPT、VE-cadherin表达水平显著降低(P<0.05)。BAY60-6583和PSB1115本身对ANGPT、VE-cadherin表达水平无显著影响,见图4。

与对照组比较,*P<0.05;与TNF-α组比较,#P<0.05
3 讨论
3.1ARDS是危重症患者常见疾病及死亡的主要原因之一,研究其发病机制及治疗措施的迫切性高。本课题组前期研究发现炎症因子(如TNF-α)刺激肺微血管内皮细胞A2BR表达升高,且在一定的时间浓度范围内具有时程及剂量依赖性[9],但A2BR对ARDS病理生理过程的影响仍有待进一步探索。本研究探讨了A2BR活化对CLP脓毒症大鼠ALI的影响,并在体外细胞水平进一步证实A2BR活化减轻TNF-α诱导的HPMECs炎症损伤,保护肺血管屏障作用:(1)A2BR活化可改善脓毒症ALI动物模型肺组织损伤情况,减轻肺水肿,提高AFC,减少炎症介质的产生和释放。(2)A2BR活化减少TNF-α刺激的原代培养HPMECs模型IL-1β分泌及ICAM-1表达,减轻肺血管内皮炎症损伤。(3)A2BR活化增加细胞间VE-cadherin表达,降低HPMECs的单层通透性;增加ANGPT表达,促进血管生成及受损内皮修复,从而对肺血管屏障起保护作用。
3.2作为异质性很强的临床综合征,基于患者临床和生物学特征进行ARDS亚型分类有助于开展个体化治疗。在基础研究中模拟ARDS/ALI的动物模型建立方法多样,脓毒症相关ALI是脓毒症并发器官损伤的常见并发症,因此本研究动物模型采用脓毒症制模的“金标准”——CLP法建立脓毒症大鼠致ALI模型,模拟了人体脓毒症合并ARDS的病理生理机制,大量炎症介质作用于基底膜血管内皮及肺上皮细胞,使其肺基底膜屏障损害,大量的高蛋白液体渗出至肺泡,出现不同程度的肺水肿,后期甚至出现肺纤维化,使肺顺应性下降,肺的气体交换功能下降,最终导致急性呼吸衰竭[10]。本研究ALI组大鼠肺组织切片可见肺泡结构紊乱,大量炎症细胞浸润,肺泡间及肺泡内可见液体渗出,同时肺W/D明显增加,AFC下降,同时肺组织内TNF-α、IL-6、IL-1β等炎症因子水平升高,证明脓毒症致ALI动物模型建立成功。
3.3腺苷A2BR是一种G蛋白偶联受体,因其对内源性配体的亲和力较低,在组织损伤(例如缺血和炎症)时产生更高浓度的腺苷从而被激活[11-12]。A2BR在炎症反应中的功能尚存在争议,如在博来霉素诱导的慢性肺损伤中肺部炎症和纤维化增加,而A2BR拮抗剂可减弱上述现象[13]。另有研究发现A2BR可以通过抑制急性炎症反应减少脓毒症死亡[14]。可能的解释是,A2BR对慢性疾病中腺苷水平的缓慢升高、急性炎症中腺苷水平的快速升高的反应不同[15]。有研究报道外源性netrin-1可通过A2BR调节炎症反应,从而对ALI产生强大的保护作用[16]。因此,腺苷A2BR是ALI治疗的重要靶点。本研究发现A2BR活化可改善脓毒症所致ALI大鼠肺组织病理损伤,减轻肺水肿,提高AFC,降低TNF-α、IL-6、IL-1β等炎症因子水平,而A2BR抑制剂可逆转上述效应,从动物层面明确在体内A2BR激活对脓毒症ALI起保护作用。
3.4肺内皮细胞炎症损伤引起的内皮通透性增加是ALI的主要病理特征之一[17]。急性感染过程中,TNF-α与内皮细胞表面的TNF-α受体结合,激活内皮信号通路并分泌炎症介质[18],这些介质诱导活性氧(reactive oxygen species,ROS)的产生、趋化因子和黏附分子分泌、抗炎介质减少以及白细胞迁移。IL-6可以将白细胞募集到肺组织中,而IL-1β可以通过诱导单核细胞和巨噬细胞加速肺损伤的过程[19-20]。除了炎症细胞因子水平升高外,在TNF-α刺激的内皮细胞中还观察到ICAM-1、血管细胞黏附分子-1(vascular cell adhesion molecule-1,VCAM-1)呈高表达[21]。本研究在体外实验中采用TNF-α刺激原代HPMECs建立细胞损伤模型,为人源细胞,与动物细胞模型相比,更能模拟人体病理生理过程。研究发现A2BR活化可减少炎症因子IL-1β分泌,下调ICAM-1表达水平,从而减轻肺血管内皮炎症损伤。
3.5VE-cadherin是黏附连接的主要成员[22]。黏附连接存在于内皮细胞及内皮细胞与基底膜之间,作为细胞通透性的重要调节因子,调节白细胞跨膜迁移和肺水肿[23]。既往有研究发现在ALI动物模型中,组织VE-cadherin表达下调[24-25]。本研究发现损伤HPMECs单层通透性增加,VE-cadherin表达水平下降,而A2BR活化可逆转该效应。但A2BR活化是否通过增加VE-cadherin表达来降低细胞通透性尚不清楚。有研究表明环磷酸腺苷(cyclic adenosine monophosphate,cAMP)的下调可降低细胞间连接蛋白的表达,从而增加血管内皮通透性[26]。据此猜测,A2BR可能通过PKA-cAMP途径增加内皮及内皮与基底膜黏附,从而降低HPMECs通透性,对肺内膜屏障起保护作用,此猜测有待进一步探索研究。
3.6本研究发现,HPMECs受损伤刺激下ANGPT表达水平下降,但A2BR活化可上调ANGPT表达水平。ANGPT是内皮细胞特异性酪氨酸激酶(endothelial-specific receptor tyrosine kinase,Tie-2)的配体,与血管内皮细胞的受体结合,诱导细胞质受体结构域的酪氨酸磷酸化,可促进血管成熟及稳定。有研究发现ANGPT不直接促进培养的内皮细胞的生长,而是对内皮细胞具有趋化作用,并参与人脐静脉内皮细胞黏附。同时,ANGPT可充当内皮细胞的凋亡存活因子,抑制培养的内皮细胞凋亡[27]。在TNF-α诱导的HPMECs损伤中,A2BR活化上调ANGPT表达,可能通过多种途径起到促血管生成作用,有助于内皮屏障的修复。有研究发现A2BR活化可经缺氧诱导因子-1α(hypoxia inducible factor-1α,HIF-1α)依赖和非HIF-1α依赖途径诱导促血管生成因子的生成[28],但内皮A2BR所介导的促血管生成效应的机制仍有待组织研究和动物研究进一步证实。
综上所述,本研究采用经CLP诱导大鼠脓毒症致ALI动物模型,从肺组织病理评分下降、肺水肿减轻、炎症因子释放减少等方面明确A2BR激活在体内可减弱脓毒症诱导的ALI。同时,在体外采用TNF-α刺激原代培养的HPMECs构建细胞损伤模型,结果显示A2BR活化可下调IL-1β、ICAM-1表达,减轻肺血管内皮炎症损伤,并通过增加细胞间黏附连接降低HPMECs的单层通透性及促进血管生成等方式促进受损内皮修复,从而对肺血管屏障起保护作用。尽管仍需要更多实验去探索A2BR对脓毒症诱导ALI保护作用的具体机制,但其对肺微血管内皮炎症损伤的抑制作用及肺血管屏障的保护作用提供了ALl潜在治疗的新靶点。
