半导体缺陷的电子结构计算方法研究进展*
2024-04-02李晨慧张陈蔡雪芬张才鑫袁嘉怡邓惠雄2
李晨慧 张陈 蔡雪芬 张才鑫 袁嘉怡 邓惠雄2)†
1) (中国科学院半导体研究所,超晶格国家重点实验室,北京 100083)
2) (中国科学院大学材料科学与光电工程中心,北京 100049)
半导体材料的掺杂与缺陷调控是实现其应用的重要前提.基于密度泛函理论的第一性原理缺陷计算为半导体的掺杂与缺陷调控提供了重要的理论指导.本文介绍了第一性原理半导体缺陷计算的基本理论方法的相关发展.首先,介绍半导体缺陷计算的基本理论方法,讨论带电缺陷计算中由周期性边界条件引入的有限超胞尺寸误差,并展示相应的修正方法发展.其次,聚焦于低维半导体中的带电缺陷计算,阐述凝胶模型下带电缺陷形成能随真空层厚度发散的问题,并介绍为解决这一问题所引入的相关理论模型.最后,简单介绍了缺陷计算中的带隙修正方法及光照非平衡条件下掺杂与缺陷调控理论模型.这些工作可以为半导体,特别是低维半导体,在平衡或非平衡条件下的缺陷计算提供指导,有助于后续半导体中的掺杂和缺陷调控工作的进一步发展.
1 引言
掺杂与缺陷调控是半导体领域的关键技术.通过掺杂引入浅能级缺陷,是调控半导体材料导电性的有效手段.相反,深能级缺陷是半导体中载流子俘获和复合中心,降低载流子浓度引起导电性降低,从而导致器件性能发生难以预测的改变.实验上观察到的器件漏电流、阈值电压漂移、载流子迁移率下降、电荷俘获、栅极电介质击穿以及温度不稳定性等器件性能的退化失效行为也均与缺陷密切相关[1–3].此外,缺陷还会影响太阳能电池的光电转换性能[4–6],光催化剂的催化效率[7,8]等.因此,实现有效掺杂和缺陷调控对半导体材料和器件性能优化至关重要.
通常,半导体材料中的缺陷种类繁多且结构复杂.在实验上,对缺陷或杂质种类和结构进行判断是非常困难的,通常需要结合多种实验手段来辨别缺陷能级对应的元素、价态、结构等信息[9–14],进而推测缺陷的种类等.而且,半导体中缺陷的低浓度以及多种缺陷经常共存的情况,又给缺陷鉴别进一步增加了难度.常见的半导体缺陷表征手段目前均不完善,例如深层瞬态光谱可以给出缺陷能级的位置,但是无法给出缺陷的结构信息,这使得实验上确定的缺陷能级找不到与之对应的缺陷[14–17];电子顺磁共振可以提供缺陷浓度、晶格环境等信息,但只能表征单电子缺陷[18].二次离子质谱法可以测量杂质浓度,但某些杂质(如氢)在低浓度下难以被检测[19].
随着计算机技术的快速发展,基于密度泛函理论(density functional theory,DFT)的第一性原理计算弥补了实验技术手段的不足,已成材料物性和新范式研究的重要手段.第一性原理计算有助于从原子尺度理解和预测材料电学、光学、磁学等诸多性质,近年来在半导体材料设计中正在发挥着越来越大的作用[20,21],尤其对半导体光电子和微电子材料发展作出了重要贡献[22].在半导体缺陷计算方面,第一性原理计算同样发挥着不可取代的作用[23],使我们在几乎不需要先验假设的情况下,对缺陷的原子和电子结构产生清晰的认识,既弥补了实验上缺陷分辨能力不足的短板,辅助解释实验现象,确定缺陷类型,同时可以提供目前实验手段无法获得的数据,完善人们对缺陷结构和性质的理解[24–26].
尽管第一性原理计算已经成为研究缺陷和杂质的有效方法,目前该领域仍然存在诸多困难,如带电缺陷的镜像电荷作用,密度泛函近似造成的带隙误差等[23].针对上述问题,目前已经发展出了相关的修正方法.在本文的第2 节,首先介绍第一性原理计算半导体缺陷的基本方法,然后介绍带电缺陷镜像电荷修正方法,具体包括Makov-Payne(MP)修正[27],Lany-Zunger (LZ)修正[28,29]和Freysoldt-Neugebauer-Van de Walle (FNV)修正[30]等经典修正,以及近期发展的自洽势(self-consistent potential correction,SCPC)修正[31]和自洽屏蔽电荷(correction scheme with screened charge density model,C-SC)修正方法[32].第3 节针对低维半导体材料体系带电缺陷能量发散的问题,介绍近期发展的低维半导体材料体系中缺陷修正方法.第4 节简单讨论了针对带隙误差修正的剪刀算符修正[33]、LDA+U[34],Heyd-Scuseria-Erzenhof (HSE)杂化密度泛函[35–37]等方法.第5 节介绍本课题组在光照非平衡条件下发展的掺杂与缺陷调控理论方法.最后,给出总结和展望.
2 缺陷计算方法与修正
2.1 缺陷计算方法
基于密度泛函理论的第一性原理方法已成功应用于研究半导体缺陷和掺杂特性,可以计算特定缺陷的一些基本性质,如缺陷形成能、离化能等.缺陷形成能定义为有缺陷的体系与无缺陷的参考体系间的总能量差,即在材料中引入一个相应点缺陷所需的能量.它描述了在材料中形成该点缺陷的难易程度.通过点缺陷在不同价态下的形成能就可以得到其缺陷浓度、载流子浓度等性质.
在第一性原理模拟中,对于带电量为q的缺陷α,其形成能 ∆Hf(α,q) 表示为
其中,Etot(α,q) 为含有缺陷 (α,q)的 超胞总能量,Etot(host) 是不包含缺陷的完美晶格超胞的总能量.在缺陷形成过程中,对于第i种元素,有ni个原子被添加到超胞中(ni<0)或从超胞中被移除(ni>0).Ei为第i种元素处于其最稳定固态或者气态时单原子能量,而µi为其参考到Ei的化学势.EF为相对于完美晶格价带顶(valence band maximum,VBM)εVBM的费米能级.为使带电缺陷的形成能有意义,需要在带电缺陷超胞与完美超胞间进行电势对齐,以使带电缺陷超胞总能与完美超胞的VBM 本征值有相同的参考点,修正项表示为
其中 ∆VPA为含有缺陷超胞与完美晶格超胞间的电势对齐.这个电势对齐通过在远离点缺陷处的原子芯能级或者静电势的变化得到.
虽然在(1)式中关于缺陷形成能的计算方法是严格的,然而在实际的缺陷计算中,特别是带电缺陷的计算过程中主要会有两方面来源的误差.第一个来源来自于有限胞下缺陷电荷与其镜像电荷间的相互作用,而另一个来源来自于半导体能隙在(半)局域泛函计算下的低估.下文将就上述存在的问题展开讨论.
2.2 镜像电荷修正
众所周知,实验上缺陷浓度一般处于1013—1018cm–3的量级,即使在重掺杂时缺陷之间也有约10 nm 的间距,这样大的间距使缺陷之间不应有相互作用.然而,在实际模拟过程中,受限于现阶段的计算能力,通常选取几百个原子的超胞中进行缺陷性质的模拟.在周期性边界条件下,缺陷电荷与其镜像电荷产生长程的库仑相互作用(图1),从而在计算出的形成能中引入相应的误差.这个误差通常在几十至几百meV 的量级,并且随缺陷带电量的增大而增大,例如,采用64 原子超胞计算的MgO 中的缺陷形成能比修正后低约800 meV,采用64 原子超胞计算的金刚石中缺陷形成能比修正后低约2.4 eV[38].由于半导体中的浅能级缺陷电离能在10 meV 的量级,镜像电荷作用引入的误差极有可能导致缺陷类型的错估,会对缺陷性质预测产生极大的误导,有限胞下缺陷形成能的镜像电荷修正因此尤为重要.
图1 周期性边界条件下的缺陷及其镜像(左)和体材料环境中的孤立缺陷(右)Fig.1.Schematic illustration of a charged defect in a finite supercell under periodic boundary conditions (left) or in an infinite large supercell (right).
为了对在周期性边界条件下这个人为引入的镜像电荷相互作用误差进行修正,目前发展了一系列的修正方法,包括MP 修正[27]、LZ 修正[28,29]、FNV修正[30]等.下面就这些修正方法的发展进行介绍.
2.2.1 MP 修正
为了修正有限胞下的镜像电荷相互作用误差,Leslie 和Gillan[39]将超胞中的缺陷电荷简化为点电荷,通过浸润在凝胶背景下的周期性点电荷所产生的Madelung 能来估算这个误差值的大小.在此基础上,Makov 和Payne[27]进一步考虑引入四极矩Q,来对缺陷电荷进行更有效的描述.最后他们将Makov-Payne (MP)修正项EMP写成了如下的数学形式:
其中αM为有限胞下的Madelung 常数,ε为静态介电常数,L为有限胞的超胞尺寸.(3)式中,正比于 1/L的一阶项对应于凝胶背景中的周期性点电荷相互作用所产生的Madelung 能,而正比于 1/L3的三阶项来自于缺陷电荷的四极矩部分与镜像点电荷间的相互作用.其余高阶相互作用有着与L更高阶次幂的减少规律,如四极矩-四极矩相互作用等,其贡献随着超胞尺寸增大迅速显小,在通常的超胞尺寸计算下可以忽略不计.
2.2.2 LZ 修正
在(3)式中,有限胞计算下唯一不直观的物理量为缺陷电荷的四极矩Q,这限制了MP 修正的实际应用.这一问题被Lany 和Zunger[28,29]进一步解决.他们给出了四极矩Q在密度泛函理论框架下的形式.他们认为,缺陷电荷主要由两部分构成,包括缺陷中心处局域的缺陷电荷,以及距离缺陷中心一定距离,由中心缺陷电荷诱导出的屏蔽电荷.他们进一步指出[28],这种屏蔽电荷在屏蔽长度lscr外拥有的数学形式,是远离缺陷的主要缺陷电荷分量,可以用来定义四极矩Q,进而确定MP 修正中的三阶项.最后,带电缺陷相互作用的LZ 修正可以表示为
其中形状因子Csh仅取决于包含缺陷的超晶胞的形状,常见体系的形状因子已经在文献[29]中给出.当介电常数ε趋向于1 时,也就是不考虑介质对缺陷电荷的屏蔽作用,LZ 修正只包含一阶项,与一阶MP 修正没有差别.而当带电缺陷周围的介电屏蔽足够大(ε≫1)时,在各向同性的超胞中(4)式简化为为一阶MP 修正项.可以看到,超胞中足够大的各向同性介电屏蔽使MP 修正相较于没有介电屏蔽时减小约1/3.最终,Lany 和Zunger[28]将LZ 修正应用到GaAs 中的,在电静势对齐的基础上加入LZ 修正项,不同超胞尺寸下的带电缺陷形成能收敛到同一值.
经过Lany 和Zunger 的工作,(3)式的MP 修正中每一个物理量都有了清晰的定义.在此基础上,Castleton 等[40]在不同带电缺陷中对MP 修正的适用性进行了充分的测试.他们发现直接通过MP 公式的一阶项进行修正无法得到准确的带电缺陷形成能,三阶项加入后得到的结果同样不准确.然而,MP 修正的数学形式却是准确的,即带电缺陷的形成能与超胞尺寸L间有
其中 ∆Hf(L→∞) 为无限大超胞中准确的带电缺陷形成能.(5)式中能量修正项分别与超胞尺寸及体积成反比,与MP 修正形式一致.按照(5)式的数学形式,将不同超胞尺寸下的带电缺陷形成能进行外推,可以得到可靠的孤立带电缺陷形成能,外推结果的误差大概在±0.05 eV[40].
按照(5)式将带电缺陷形成能外推时,需要对超胞的3 个方向同时进行缩放.然而很多情况下由于计算能力的限制,3 个方向同时进行缩放的作法只能给出少量的数据点.拟合过程中数据点的不足为最后的外推结果带来很大的不确定性.为了解决这个问题,Hine 等[41]提出同时改变超胞尺寸和形状可以提供更多的数据点,从而大幅减小这种不确定性.他们考虑离子晶体中能较好地被点电荷描述的不同带电缺陷,此时一阶MP 修正已经足够.他们计算了尺寸和形状皆不同的超胞中的Madelung势νM=α/L,并将不同超胞中的带电缺陷形成能按照与νM线性外推到νM=0,从而得到准确的孤立带电缺陷形成能.他们对Al2O3中不同带电缺陷的测试显示,这个修正方法在尺寸和形状皆不同的超胞中给出了相一致的形成能[41].此外,他们进一步考虑各向异性介电屏蔽的影响,通过介电张量将各向异性电荷屏蔽效应纳入Madelung 势的计算中[42].对具有高度各向异性介电张量的单斜晶系Li2TiO3中不同带电缺陷的测试显示,不同超胞中修正后的带电缺陷形成能收敛到准确值的0.1 eV左右.
2.2.3 FNV 修正
尽管基于超胞缩放的形成能外推对镜像电荷相互作用误差展现出了强大的修正能力,然而对于每一种缺陷都要在不同超胞尺寸下进行计算使这种修正方法变得繁重且昂贵.而与超胞缩放相比,一种更直接的方法是在有限胞下直接计算镜像电荷相互作用能,从而导出这个误差.为此,Freysoldt等[30]发展了著名的FNV 修正方法.FNV 方法将周期性边界条件下缺陷引入的电势进行拆分:
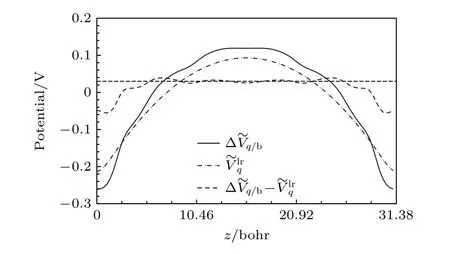
图2 立方GaAs 超胞(3×3×3)中 的电势分布,其中分别为 缺陷的 总势场、长 程势和短程势,水平虚线指示潜在的电势对齐项,缺陷和镜像分别位于z=0 和z=31.38 Bohr 处,图片引用自参考文献[30] (版权属于美国物理学会)Fig.2.Potentials for a in a 3×3×3 cubic GaAs supercell.The and are respectively the total,long-range and short-range part,of the defect electrostatic potential.The potential alignment term is indicated by the dashed line.The defect is located at z=0 Bohr with a periodic image at z=31.38 Bohr.Reprinted with permission from Ref.[30],copyright 2009 by the American Physical Society.
考虑到带电缺陷的长程作用势在距离缺陷较远处变化缓慢,与缺陷的细节关联较弱,因此可以通过简单的模型电荷qd(如点电荷、高斯电荷等)以及宏观介电常数,构建周期性边界条件下的泊松方程来求解长程势.而在缺陷附近,由于缺陷周围复杂的短程屏蔽及明显的结构弛豫,缺陷中心附近势场变化较快(图2),短程势难以用简单的模型电荷以及宏观介电常数来构建.因此,FNV方法直接通过从DFT 计算得到的缺陷电势(带电缺陷与中性缺陷的电势差值)减去模型电荷电势提取短程势.短程电势在超胞内快速衰减,在拟合势场时,要求短程势在远离缺陷的位置出现变化平缓的“电势平台”.这个“电势平台”的高度即为电势对齐项C,其本质来源于带电缺陷与中性缺陷超胞的势场在进行相减时所需要的电势对齐.
式中第1 项来源于周期性排列的模型电荷引起的Madelung 能:
其中n=–q/Ω为背景凝胶的电荷密度.而第2 项来源于短程势的贡献:
Freysoldt 等[30]将得到的FNV 修正项用于修正GaAs 中不同价态Ga 空位的形成能,他们的测试结果显示在不同的超胞尺寸下,所计算出的缺陷形成能收敛到了同一值附近,显示了FNV 修正的有效性.
尽管FNV 修正展现出了直观有效的物理图像,然而在运用到某一些半导体材料时仍然存在一些困难.一方面,FNV 修正采用沿某一方向的平面平均静电势作为电势对齐的标准,这在计算具有较小结构弛豫的点缺陷是有效的.然而,在一些离子性较强的半导体中,带电缺陷的形成在位于缺陷较远的地方仍能引起明显的结构弛豫,此时在沿特定方向上的“电势平台”难以被观测到,电势对齐项难以被直接求出.而另一方面,FNV 修正采用宏观介电常数对长程屏蔽效应进行描述.但这在电势屏蔽各向异性较强的体系中(如层状半导体等)变得不再适用.为了解决以上两方面的限制,Kumagai和Oba[43]对原本的FNV 方法进行了改进,一方面,他们不再采用远离缺陷的“电势平台”的高度,而是参照Lany 和Zunger[28,29]的做法,用排除掉缺陷周围原子的平均原子局域势作为电势对齐的标准,从而使FNV 修正在具有大弛豫结构的缺陷中能够得到应用.另一方面,他们在构建长程势时充分考虑了晶体中的各向异性屏蔽,从而将FNV 方法扩展到各向异性材料.他们将改进后的FNV 方法应用到了层状β-Li2TiO3中不同带电缺陷计算中,结果显示在不同尺寸的超胞中修正后的结果能够收敛到同一值,显示出他们所改进的FNV 方法的有效性[43].然而,改进后的FNV 方法依然依赖于模型电荷对长程势的构建,这就说明FNV 方法对一些十分局域的缺陷有很好的修正能力,而对一些非局域的缺陷可能并不是特别适用.
2.2.4 SCPC 修正
经典的修正方法忽略了周期排列的镜像电荷以及背景电荷对单粒子能级的影响,因此可能会影响体系总能的计算,导致错误的结果.尤其是在对具有负电子亲和势的表面模型计算过程中,背景补偿电荷会在带隙中引入虚假的真空态,导致电子的错误占据[31](图3(a)).针对这一问题,Da Silva[31]最近提出的自洽势场修正方法(SCPC),在自洽求解Kohn-Sham 方程时把周期边界条件下缺陷电荷的势场修正到开放边界条件下缺陷电荷的势场,以此修正缺陷的镜像相互作用.SCPC 修正采用带电缺陷体系与参考体系(无缺陷的host 体系或者中性缺陷超胞)的电荷密度差作为缺陷电荷 δρ=ρchg-ρref,分别计算该电荷在周期性边界条件和开放边界条件下的势场Vper和Viso,根据Vper和Viso确定体系势场的修正量Vcor=Vper–Viso并在哈密顿量中引入Vcor,得到无缺陷相互作用的哈密顿量,通过不断的自洽迭代修正缺陷电荷分布 δρ以及缺陷势场Vper.由于SCPC 修正在哈密顿量中消除了来自于镜像电荷的伪库仑相互作用,因此可以同时修正KS 单电子能级以及波函数.
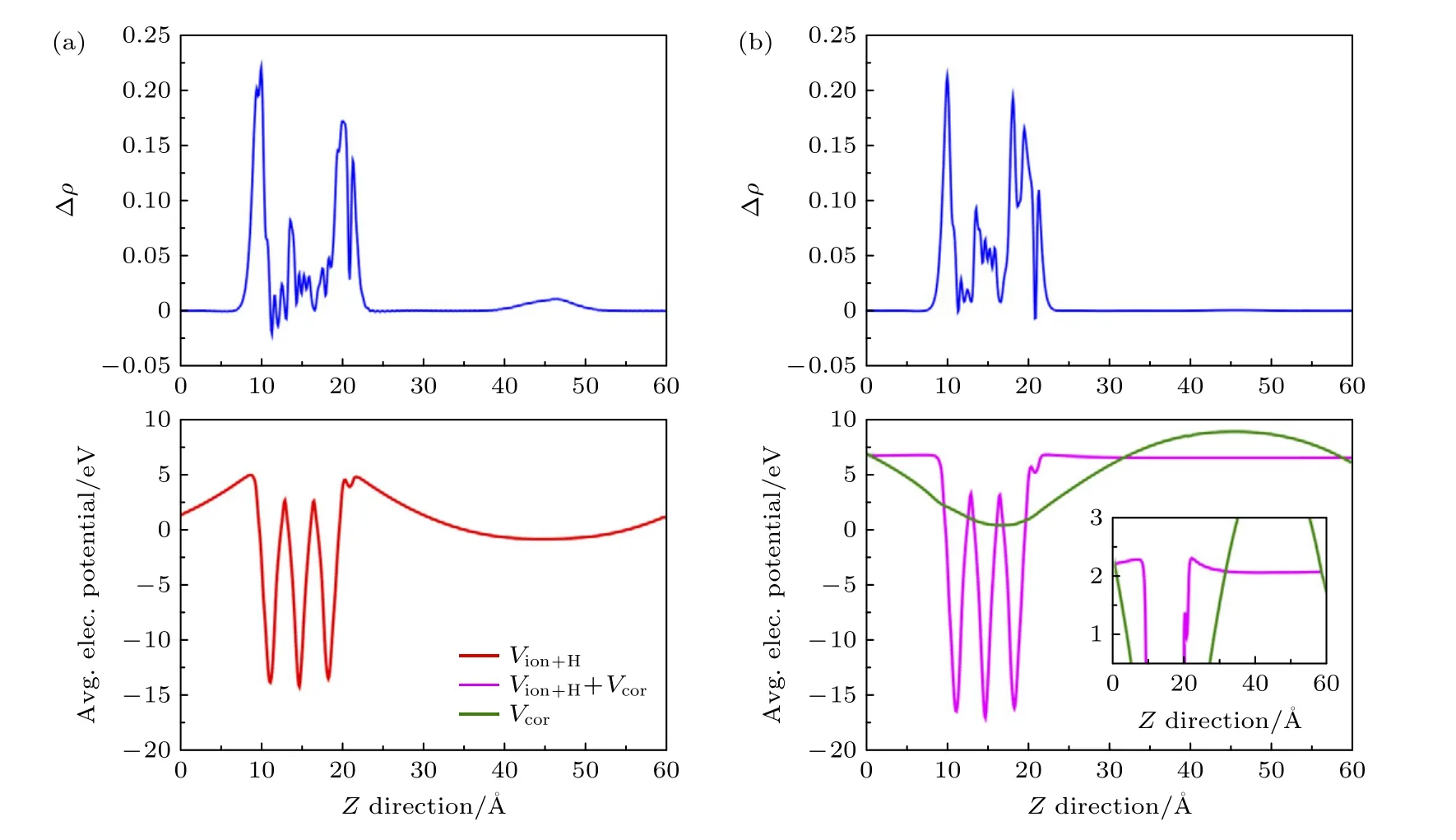
图3 锐钛矿相TiO2(001)片层表面 沿表面法向的面平均电荷和平均静电势分布 (a)未经过SCPC 修正;(b)经过SCPC修正,红色和玫红色曲线分别代表修正前后离子与电子的总势场,绿色曲线代表修正势,经过SCPC 修正后,真空内的势场升高,电荷分布消失;图片引用自参考文献[31] (版权属于美国物理学会)Fig.3.Planar average of the extra charge and the electrostatic potential along the surface normal for an molecule on the surface of an anatase-TiO2 (001) slab,without (a) and with (b) the SCPC correction.Reprinted with permission from Ref.[31].Copyright 2021 by the American Physical Society.
此外,SCPC 还能够修正背景补偿电荷导致的虚假真空态占据.对于带负电荷的类受主缺陷,分布在真空的补偿电荷降低了真空区域中的电子态,当其低于被占据的缺陷能级时,会导致真空态的非物理占据.如图3(a)所示,未经SCPC 修正时,锐钛矿相TiO2(001)片层表面吸附的模型真空中的静电势明显低于表面的静电势,在真空部分有明显的电荷分布,经过SCPC 修正后,补偿电荷的影响被消除,真空中势场接近材料表面静电势,且沿表面法向无明显变化,真空中的电荷分布消失(图3(b)),说明SCPC 可以修正补偿电荷在真空导致的虚假势场,这对包含真空层的低维材料缺陷计算有重要的应用价值.然而,SCPC 在修正过程中还是依赖宏观介电常数或者介电剖面的计算,不仅使计算过程更加复杂,还会由于人为设定介电常数引入计算误差.
2.2.5 屏蔽缺陷电荷C-SC 修正
为了降低经典修正方法及SCPC 自洽势修正中人为设定介电常数对缺陷能量计算的影响,Suo等[32]最近提出了基于DFT 自洽电荷的屏蔽缺陷电荷C-SC 的修正方法,他们考虑了缺陷周围束缚电荷的极化响应,认为其中包含了介质对缺陷的屏蔽作用,因此将体系总能展开成屏蔽缺陷电荷密度(图4(a),(b))和无屏蔽的缺陷电荷密度(图4(c))相关的函数时,不需要额外设定宏观介电常数,整个过程可以在自洽场计算中进行求解.他们发现缺陷电荷(带电缺陷与中性缺陷体系的电荷密度差,图4(a))可以分解成核心电荷(图4(b))以及接近常数的背景电荷,其中有限超胞中核心电荷快速收敛至孤立体系的核心电荷而背景电荷CN与超胞的体积成反比通过比较周期体系和开放边界条件下体系的总能进一步推导出镜像电荷的相互作用:
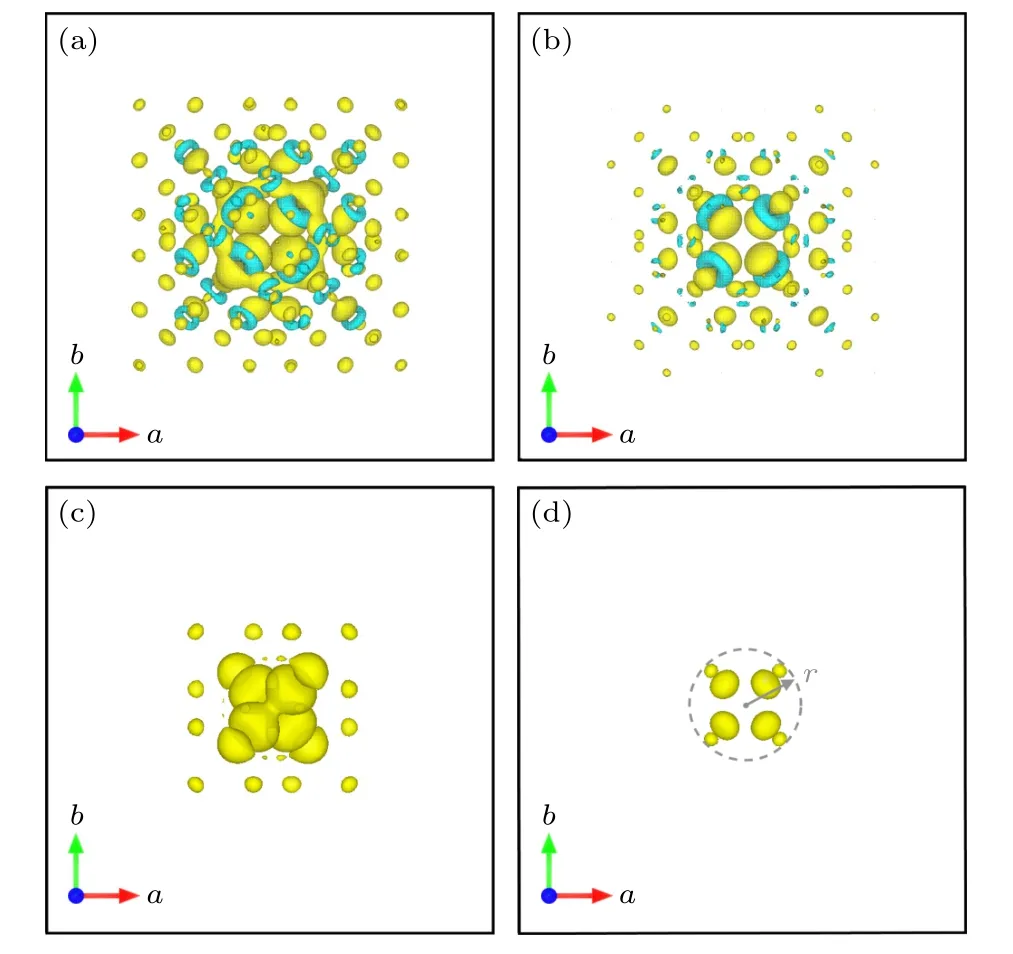
图4 金刚石超胞(4×4×4)中的 的缺陷电荷密度 (a)屏蔽缺陷电荷密度 ;(b)屏蔽缺陷电荷的核心电荷密度;(c)无屏蔽的缺陷电荷密度 ρd=|φd|2,其中 φd 为缺陷态的 波函数;(d) ρd(r)/ε∞ (ε∞=5.62),图片引 用自参考文献[32] (版权属于美国物理学会)Fig.4.Defect charge distribution for in a cubic 512-atom diamond supercell (a=14.13 Å): (a) The screened defect charge density ;(b) core defect charge density;(c) unscreened defect charge density ρd=,where φd is the wave function of the defect state,and (d)ρd(r)/ε∞ (ε∞=5.62).Reprinted with permission from Ref.[32],copyright 2020 by the American Physical Society.
该修正方法直接从自洽场计算中提取保留了屏蔽作用的缺陷电荷分布(图4(a)),因此无需再通过宏观介电常数来描述屏蔽作用,是一种更精确的带电缺陷能量修正方法.对缺陷体系的介电常数接近宏观介电常数常数的情况,例如带电量低的缺陷(单个电荷)和高对称性缺陷,屏蔽缺陷电荷修正法与FNV 方法修正结果非常接近.而对某些带电量较高的缺陷,例如MgO 中的缺陷,其中存在非线性屏蔽效应使得介电常数明显偏离宏观介电常数[32],基于自洽计算的缺陷屏蔽模型可以很大程度上降低人为设定宏观介电常数导致的修正误差.
3 低维半导体材料中的缺陷计算
以上的讨论都聚焦在三维半导体材料体系,然而在低维结构中传统的带电缺陷修正方法遇到了更严峻的挑战.虽然,同样可以仿照传统三维半导体中的做法,同时在3 个方向缩放超胞,将带电缺陷形成能按照MP 修正的形式外推到无穷大的超胞中,从而得到其准确值.然而,因为需要保持一定厚度的真空层,缩放后的超胞很可能在真空层方向有很大的超胞尺寸,从而使这种计算变得费力且昂贵.另一方面,也可以将FNV 方法或者SCPC方法应用到低维半导体中[31,44].FNV 方法与SCPC方法都依赖于通过静态介电常数构建长程势,因而需要提取真空层方向的介电剖面.额外的介电剖面提取过程一定程度上损害了这两种方法的简便直接性.
另外,低维结构进行建模时需要包括一定厚度的真空层,为了尽量减小层与层之间的相互作用,人们甚至希望真空层厚度Lz→∞.然而,在传统的凝胶模型中,凝胶背景电荷虚假的分布到了真空层中,从而造成随着Lz增大而发散的超胞库仑能,最终使计算得到的带电缺陷形成能随着真空层厚度而发散(图5(a),(b))[45].这个发散行为为以往具有任意选取真空层厚度的有限胞中带电缺陷的计算结果带来很大的误差[44].另一方面,对于受主缺陷而言,真空层中人为设置的凝胶电荷还可能造成缺陷电荷在真空能级上的错误占据[45],从而导致错误的带电缺陷计算结果.为了解决在传统凝胶模型中低维半导体中的带电缺陷形成能随真空层厚度发散的问题,人们提出了很多方法.例如,Wang等在二维情况下将离化能展开为面内超胞面积S和真空层厚度Lz的泰勒级数,并在极端超胞尺寸下保留展开中的重要项[45].最终他们得到了二维材料中的离化能随超胞尺寸的缩放关系为: IE(S,Lz)=其中α和β为拟合系数,而IE(S,Lz)和IE0分别为有限胞与无限大胞中的离化能.将具有不同超胞尺寸(S,Lz)中的离化能按照上式进行外推即可得到孤立缺陷离化能IE0.他们将这个外推方法应用到了单层BN 中的多种点缺陷,结果显示在不同尺寸的超胞中按照这个方法进行修正后可以得到收敛的离化能.同样他们也发展了具有一定厚度二维半导体中的带电缺陷计算结果的外推方法[46],此时他们发现正比于1/S的项不再能被忽视.尽管这个外推方法直接且易于操作,但它依赖于不同超胞下的计算结果,计算过程依旧繁琐,而且外推过程如何应用到界面等更复杂的低维体系中没有得到解答.另外,Richter 等[47]将电离电荷放到局域在低维结构的一些人为态上可以直接避免随着真空层发散的带电缺陷形成能的出现,这个过程可以通过修改结构中的原子核数进行.然而,这种直接修改原子核数的做法缺乏物理层面的合理解释.
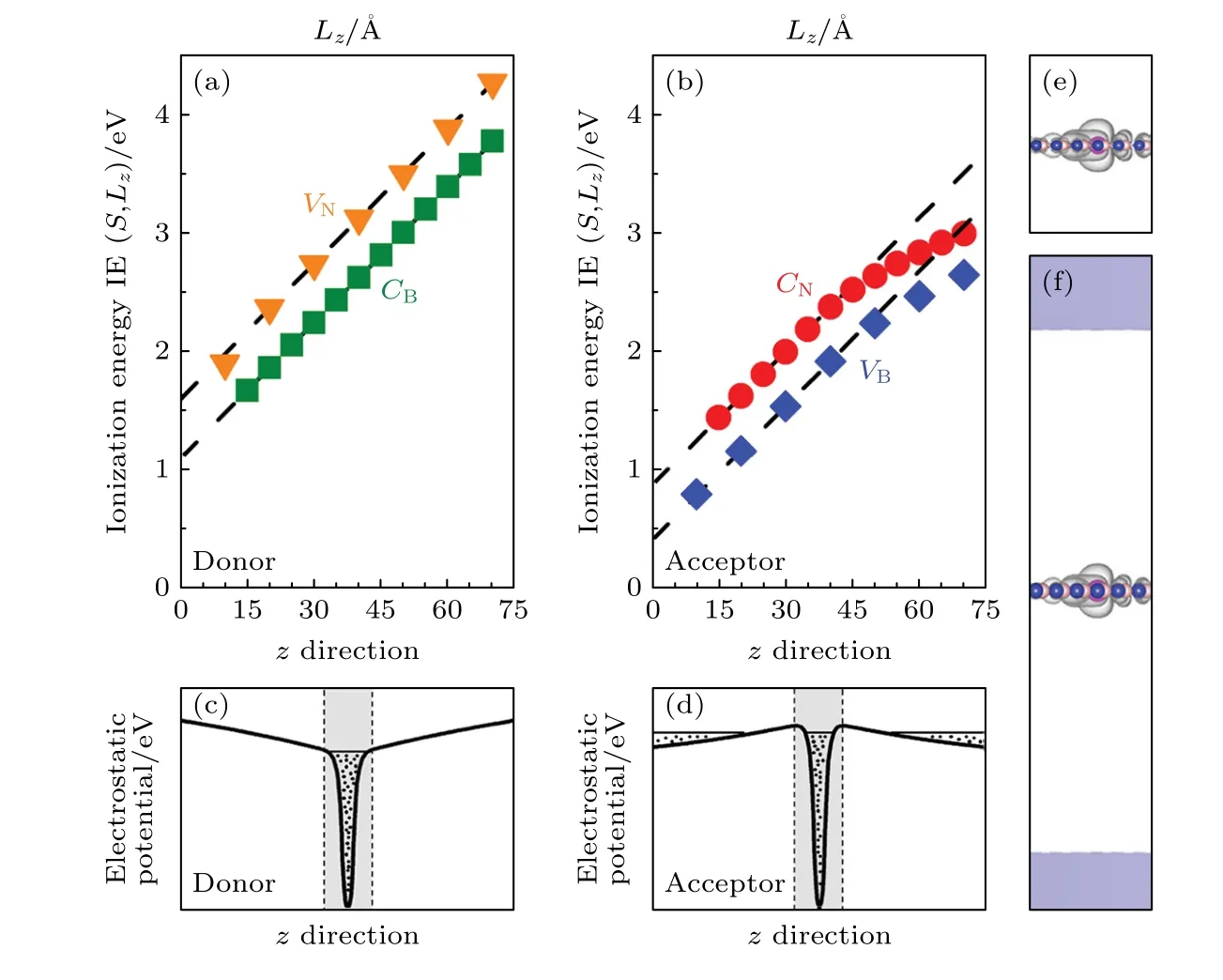
图5 二维BN 的(a)施主(CB 和VN)及(b)受主缺陷(CN 和VB)的离化能与超胞z 方向长度的关系;(c)和(d)分别是施主和受主缺陷在z 方向的静电势分布,水平直线代表费米能级的位置;(e),(f)分别是采用Lz=20 和70 Å的超胞计算的 缺陷的电荷密度分布,(f)中的阴影部分表示电子在虚假真空态的占据.图片引用自参考文献[45] (版权属于美国物理学会)Fig.5.Calculated ionization energies of (a) donors,CB and VN,and (b) acceptors,CN and VB in 2D BN,as a function of cell le ngth in z direction (Lz);(c),(d): schematic illustration of the corresponding electrostatic potentials;(e),(f) acceptor state in at different Lz=20 and 70 Å,respectively.The shade areas at the top and bottom of panel (f) are the calculated defect states unphysically delocalized into the vacuum.Reprinted with permission from Ref.[45] .Copyright 2015 by the American Physical Society.
总之,目前对低维半导体体系带电缺陷的计算大多依赖于人为的后处理或者修正,缺乏一个对缺陷电离过程的直观物理理解.物理上,缺陷电子电离过程对应于缺陷电子(空穴)转移到CBM(VBM)的一个真实过程,应该是不随体系维度结构的变化而改变,因此,凝胶模型在低维半导体缺陷计算上的表现难以让人满意.人们需要一个对任意维度半导体中带电缺陷计算的普遍适用的理论方法.
为此,本课题组提出了转移真实态的模型(transfer to real state model,TRSM)[48].当考虑一个施主缺陷的真实电离过程时,电子从缺陷态被激发到母体材料的CBM 态上,如图6(a)所示.电离载流子的电荷分布不应由三维空间中均匀分布的凝胶电荷描述,而应由真实CBM 态的电荷分布描述.基于此,TRSM 下的带电缺陷形成能可写为
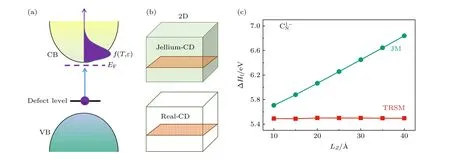
图6 (a) 转移真实态模型示意图,电子从缺陷电离到导带,导带的电子占据服从费米-狄拉克分布;(b)二维结构缺陷模型在凝胶模型和转移真实态模型电离出的电荷密度分布示意图,绿色阴影代表凝胶电荷分布 (jellium-CD),橙色区域点状标记代表真实态的电荷分布 (real-CD);(c) 凝胶模型(JM)和TRSM 修正的二维BN中 的缺陷形成能与层间距Lz 的关系.图片引用自参考文献[48] (版权属于美国物理学会)Fig.6.(a) Schematic plot of the TRSM model,where the electrons excited from the defect state to the conduction band minimum(CBM) follow the Fermi-Dirac distribution;(b) schematic plots of the jellium charge distribution (jellium-CD) and real state charge distributions (real-CD) in a model with a 2D layer (in orange).The jellium and real charges are represented by the green and dotted orange area respectively;(c) formation energies of in BN monolayer corrected by the jellium model (JM) and TRSM respectively,as functions of layer separation Lz .Reprinted with permission from Ref.[48],copyright 2020 by the American Physical Society.
4 DFT 带隙修正
半导体能隙计算的合理性对缺陷性质的模拟至关重要.然而,密度泛函理论中的交换关联能没有确切形式,通常会采用诸如局域密度近似(LDA)和广义梯度近似(GGA)等(半)局域泛函.这些泛函在计算大多数物质的结构、能量等方面可以给出相当准确的结果,但在计算半导体材料时仍然具有明显的不足,LDA/GGA 泛函计算下半导体的带隙值会被严重低估[49,50].例如,LDA/GGA 对ZnO和SnO2带隙的低估分别达到了70%和75%[51,52],计算CdO 和InAs 时带隙甚至为负值[52,53].这个对带隙值的低估一般是由于多电子自相互作用产生的[49,50,54–56].考虑体系在N电子的基础上有δn个电子的变化,体系的 Hartree 项变为
(12)式中第1 项仅为体系变化前的Hartree项,第2 项与δn成线性关系,表现为从N→N+δn变化过程中体系能量呈现线性变化.第3 项为虚假的自相互作用项,由于这项的存在,能量随电子数的变化偏离了线性行为增加了开口朝上的二次项贡献(图7).向下凸起的能量变化曲线使电子更倾向于分数占据而不是整数占据,这也是在一些情况下(半)局域泛函在预测例如极化子等局域电子态时会失效的原因.考虑带隙值的计算:
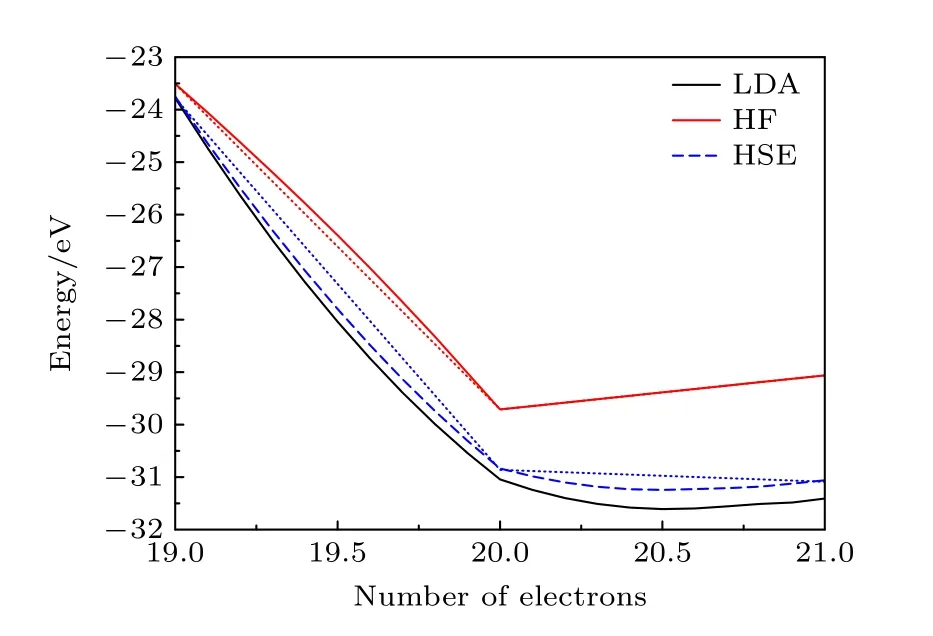
图7 由LDA、Hartree-Fock 理论和HSE 混合泛函得到的Si4H4 团簇的总能量与电子数量的关系,点线表示精确解.图片引用自参考文献[23] (版权属于美国物理学会)Fig.7.Total energy of a Si4H4 cluster with respect to the number of electrons,for the LDA,Hartree-Fock theory,and hybrid functional (HSE).The dotted straight lines denote the ideal exact solutions.Reprinted with permission from Ref.[23],copyright 2014 by the American Physical Society.
可以看到自相互作用项使能量随电子数的变化经过N电子时一阶导的增大而大幅下降(图7),由此造成了对带隙值的严重低估.对带隙描述的不准确极大地限制了人们对于缺陷,特别是带电缺陷的性质预测能力.因此,需要对DFT 下半导体带隙的计算进行修正.
半导体带隙修正经过多年发展可以通过多种手段来实现,例如,一种简单的方法是通过经验修正赝势进行带隙修正[57],该方法应用于浅能级缺陷计算已取得较好效果[58,59].下文主要介绍早期的剪刀算符修正,LDA+U方法以及目前应用最多的HSE 杂化泛函等几种典型的带隙修正方法.
关于半导体带隙修正,最早发展的方法为剪刀算符.剪刀算符核心在于缺陷态可以完备地投影到导带和价带布洛赫态上,当刚性移动导带位置和实验带隙值对齐时,缺陷态根据其导带布洛赫态的比例获得等比例的移动[60,61].但很多时候这种直接移动能带的方法显得有些粗糙.之后Zhang 等[60]发展了基于剪刀算符的外推方法,他们的思想在于随着外参数λ 变化,所计算出来的带隙值Eg(λ)和形成能ΔHf(λ)间呈现连续变化,通过Eg(λ)和ΔHf(λ)间的一阶依赖近似可以将ΔHf(λ)外推到Eg(λ)=时的值.但是如何选取外参数λ 使其具有真实的物理意义没有一个很好的答案,因此基于这种方法的计算结果仍然存在很大的不确定性[23,60,62].
一种具有真实物理意义的外参数λ 的选取方式为调节半芯d 态上的库仑排斥能U,即LDA+U的方法.半芯d 态在GaN,InN,ZnO 等氮化物,氧化物半导体中广泛存在.在(半)局域泛函的描述下,半芯d 态的局域性被低估,造成其在能带中的位置过高.而当在半芯d 态上施加额外的库仑排斥能U时,其波函数的局域性得到一定程度的恢复.此时半芯d 态受到更强烈的原子核吸引,导致其在能带中的位置被拉低约U/2.以ZnO 为例,一个更低的半芯d 态可有效地减小d-p 耦合,从而使VBM态的能量降低;并且更局域的半芯d 态可以使外层s 态得到更好的屏蔽,从而使CBM 态的能量提高[51,63].从这两个方面,被DFT 低估的带隙值得到一定程度的修正.然而,LDA+U的方法难以适用于Si 等缺少半芯d 态的半导体[64].另一方面,U值的选取很大程度上依赖于经验方法,具有一定的任意性.尽管Cococcioni 和de Gironcoli[65]发展了基于线性响应理论的第一性原理计算U值的方法,但是由于LDA+U并不能从根本上修正(半)局域泛函导致的多电子自相互作用,因此在合理的U值范围内修正后的带隙值依然低于实验值[66].
除了以上外参数调节的方法,半导体带隙值的修正还可以直接从修正交换泛函开始.Becke[67]最先提出,为了改善被传统DFT 计算低估的长程屏蔽,可以在(半)局域泛函中添加一定比例的非局域Hartree-Fock(HF)交换泛函的成分.之后Muscat等[68]发现这种杂化泛函相较于(半)局域泛函对固体带隙的预测能力得到显著改善.这是因为HF 交换泛函下能量随着电子数的变化同样偏离线性行为,然而与(半)局域泛函不同的是,变化曲线呈现向上突起的特征,如图7 红色曲线所示.通过在(半)局域泛函中混入非局域HF 交换势,能量随电子数的变化可以一定程度上恢复线性变化,从而更加准确地预测带隙值.在这个思想的基础上,Heyd 等[35,36]考虑到固体中的介电屏蔽,为混入的Hartree-Fock 交换泛函添加了相应的截断,从而大大简便了其在固体中的应用,这个改善后的泛函即为目前半导体缺陷计算最常用的HSE 杂化泛函.一系列的半导体带隙计算结果显示,当HF 泛函混入比例为α=25%,截断参数μ=0.2 Å–1时,HSE 泛函能对大多数半导体的带隙值有很好的预测[69,70].少数屏蔽性质比较特殊的半导体或绝缘体,例如MgO 等在使用HSE 泛函时需要进一步对这两个参数进行调整.当半导体的带隙值得到修正后,HSE 泛函对缺陷性质的预测也十分出色.对于金刚石的NV–中心,使用HSE06 计算3A2→3E跃迁为2.21 eV[71],与实验值2.18 eV[72]非常接近.Komsa 和Pasquarello[73]通过混合泛函计算GaAs内As 反位缺陷的(+2/+1)和(+1/0)转变能级与实验误差在0.2 eV 以内[74].然而,HSE 泛函的计算速度是限制其应用的瓶颈,由于Hatree-Fock 交换需要计算四中心积分,在相同电子总数的情况下,HSE 计算速度比LDA 慢至少一个数量级[75].此外,HSE 等混合泛函会影响缺陷波函数的局域性,计算类氢缺陷时会高估转变能级的深度[76],导致预测的缺陷性质同实验结果存在较大误差,因此在计算非局域性较强的浅能级缺陷时需要仔细筛选截断长度等计算参数.同时,基于更高精度的缺陷计算方法更值得期待,如GW 方法.
5 非平衡态光照条件下的缺陷计算
目前,缺陷和掺杂理论主要基于热力学平衡条件,该条件下存在一些固有的掺杂限制,如固浓度极限和本征自补偿缺陷等[77,78].具体而言,在热力学平衡条件下,系统具有单一的费米能级,无法在抑制本征自补偿缺陷的同时增加自由载流子浓度.然而,在非平衡生长条件下,电子和空穴的费米能级得以解耦,如图8(a)所示,半导体中的平衡费米能级EF在光照下分裂成两个准费米能级,这扩大了调控范围和自由度,有望克服平衡条件下的限制,突破掺杂瓶颈.因此,发展系统的热力学非平衡缺陷和掺杂理论对于理论和实际应用都具有重要意义.
以往的实验发现,在半导体生长过程中施加光照可以有效提升多种化合物的掺杂效率,包括CdTe,GaN 和ZnX(X=O,S,Se,Te)[77,80–85].尽管研究者们已经提出了多种计算方案来研究光照对半导体掺杂特性的影响,然而对这些影响的物理认识仍然相当有限.近期本课题组提出了一种自洽的“双费米库(TFR)”模型[79]: 由于平衡费米能级通常被视为费米存储库(reservoir),相应地,首先将非平衡态下的两个准费米能级类比为两个准费米库(用Res.C 和Res.V 表示).考虑到这两个准费米库都会对电子激发到导带和空穴激发到价带有贡献,基于经典玻尔兹曼统计,定义了准费米能级Efc对于电子激发到导带(Efv对于空穴激发到价带)的贡献权重因子这里的 ∆Ef=Efc-Efv是准费米能级差,常用于衡量系统偏离平衡态的程度.基于这个权重因子,进一步得到了从这两个费米库激发的载流子浓度表达式,并定义了缺陷有效费米能级,获得相应的缺陷形成能和缺陷浓度公式.最后,通过自洽求解电中性条件和非平衡稳态条件,便可以获得两个准费米能级的值,从而获得其他描述系统的物理参数,包括载流子浓度、缺陷形成能和缺陷浓度等.
基于此模型,阐明了光照提升半导体掺杂效率的基本机制: 施加光照引入的光生载流子会使Efc和Efv两个准费米能级分别向CBM 和VBM 移动.因而相应地,施主和受主的有效费米能级也分别向CBM 和VBM 移动,从而同时增大了施主和受主的形成能,减少了缺陷浓度.但由于费米能级的偏移量与相应载流子浓度成反比的关系,即 |dEF|=(N=n或p),因此施主和受主的有效费米能级的移动是不对称的.对于n 型掺杂EfA的偏移量远大于EfD.而对于p 型掺杂,EfD偏移量远大于EfA.这使得补偿施主的减小量明显大于目标缺陷浓度的减小量,从而增大了系统中的多数载流子浓度.因而施加光照可以提高半导体材料的有效掺杂效率,这与以往的实验观察结果一致[85].图8(c),(d)展示了在非平衡稳态光照条件下,p 型掺杂的GaN:Mg 体系中MgGa及补偿缺陷VN的浓度和体系中载流子浓度与光产生率G的关系.可以看到,随着光产生率G增大,缺陷浓度都逐渐减小,且在每个G下,补偿缺陷VN浓度的减少比受主缺陷MgGa更为显著;另外,随着光产生率G增大,计算得到体系中的电子和空穴浓度均得以增大,这符合物理直觉,且与以往实验的结果(光照强度的增加导致自由载流子浓度增加)相吻合[85].此外发现,当撤掉光照时,经过光照的系统相对于未经过光照的系统仍具有更大的空穴浓度增量.该研究不仅揭示了通过光照增强半导体材料掺杂效率的内在物理机制,而且为克服半导体中的掺杂限制提供了一个普适的途径.
6 总结与展望
本文概述了基于密度泛函理论的第一性原理缺陷计算的基本理论和相应的修正方法,主要涵盖以下几个方面: 1)介绍了常用的半导体缺陷计算方法(即超晶胞法),并总结了为解决带电缺陷计算中由周期性边界条件引入的有限超胞尺寸误差而发展的几种关键的镜像电荷修正方法,具体包括MP 修正、LZ 修正、FNV 修正、SCPC 修正、以及屏蔽缺陷电荷C-SC 修正;2)讨论了传统凝胶模型中低维半导体中带电缺陷形成能在真空层厚度上发散的问题,以及本课题组基于对缺陷电离物理过程的理解,提出的转移真实态模型,并指出它是一个与维度无关的带电缺陷计算模型;3)阐述了几种常见密度泛函在预测半导体带隙值时的准确度,以及相应修正方法的提出和发展,包括最早的剪刀算符、调节半芯d 态上的库仑排斥能的LDA+U方法,以及杂化泛函方法;4)介绍了用于研究光照对半导体掺杂特性影响的自洽双费米库模型,并揭示了通过光照增强半导体材料掺杂效率的内在物理机制.
尽管基于密度泛函理论的第一性原理缺陷计算方法取得了显著进展,但仍然面临一系列困难与挑战: 1)在缺陷性质研究方面,目前主要关注缺陷的形成能、浓度及离化能等,而对于缺陷的散射与俘获特性、多声子非辐射跃迁及缺陷动力学等其他一些重要性质的研究和理解仍然较为缺乏;2)对于非周期性体系,如无序合金、非晶等,由于其晶格缺乏平移对称性,需要对大量不同点位的缺陷性质在给定温度下进行统计平均,使得第一性原理缺陷计算面临计算量巨大的挑战[86,87],因此,需要在保持计算准确度的前提下发展更为快捷的缺陷计算方法,如机器学习算法;3)对于非平衡条件(如压强、变温、离子注入、电场等)对半导体掺杂特性的影响,目前的研究仍处于初级阶段,需要更深入的实验验证和理论探讨;4)本文介绍的缺陷计算方法主要针对半导体中的点缺陷,然而在实际材料中除了点缺陷还存在其他缺陷类型,包括线缺陷、面缺陷及多种缺陷复合等.由于这些缺陷类型较复杂,计算成本也较高,目前人们对它们的特性尚缺乏深入认识.随着更先进的计算模型与方法的提出、计算能力的不断提高以及理论与实验更完美的结合,这些困难与挑战有望得以解决,从而推动半导体物理科学的进一步发展.
