整体式钴基复合金属催化剂微波催化燃烧甲苯特性
2024-03-28赵思蕊卜龙利代菁雯黄思宁罗梦垚王嘉乐西安建筑科技大学环境与市政工程学院西安70055西北水资源与环境生态教育部重点实验室西安70055陕西省环境工程重点实验室西安70055
赵思蕊,卜龙利,2,3*,代菁雯,黄思宁,罗梦垚,刘 楠,王嘉乐(.西安建筑科技大学环境与市政工程学院,西安 70055;2.西北水资源与环境生态教育部重点实验室,西安 70055;3.陕西省环境工程重点实验室,西安 70055)
工业VOCs 排放源包括化工、工业涂装、包装印刷等行业,其排放量约占全国人为源VOCs 排放总量的55.5%[1].因此,工业VOCs 防控是我国“十四五”时期大气污染治理的一项重点工作.催化燃烧作为一种治理VOCs 的有效技术,具有起燃温度低、节能、处理效率高、无二次污染以及适用范围广等优点[2].与传统电加热无选择性热传导的加热方式相比,微波加热对于可吸波型催化剂具有选择性加热的特点.在微波辐射作用下,催化剂表面负载的活性组分偶极极化,由于分子间的摩擦、碰撞与转动,进而将电磁能转化为热能[3],使得催化剂表面某些微区温度高于催化剂的整体宏观温度[4],即热点效应.热点效应与选择性加热是微波辐射诱导催化反应较之传统电加热方式催化效率提升的关键,能够产生温度更高的热活性位点,这些“热点”可以降低反应的活化能,使VOCs 分子键更易于断裂,催化反应更加易于进行[5]. Yi 等[6]制备了MnO2(Ac)催化剂,发现催化温度150℃时,微波加热下甲苯的转化率比电加热下提高了59%,矿化率提高了36%,反应活化能降低了88.3KJ;卜龙利等[7]制备了Cu-Mn-Ce/TiO2-分子筛催化剂,发现在微波加热方式下,床层温度145℃时甲苯的降解率达94%,而采用电炉加热至床层温度265℃时,甲苯的降解率仅为77%.研究发现,利用微波催化燃烧技术可以实现在较低的床层温度下完成甲苯的氧化降解[8-9].
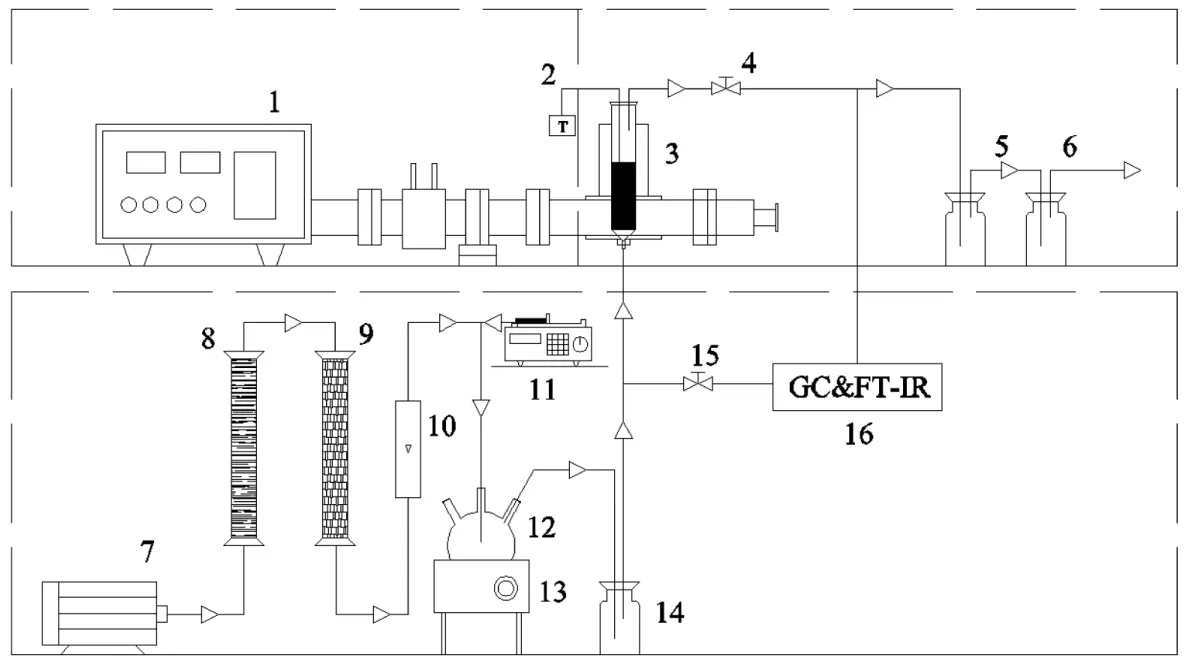
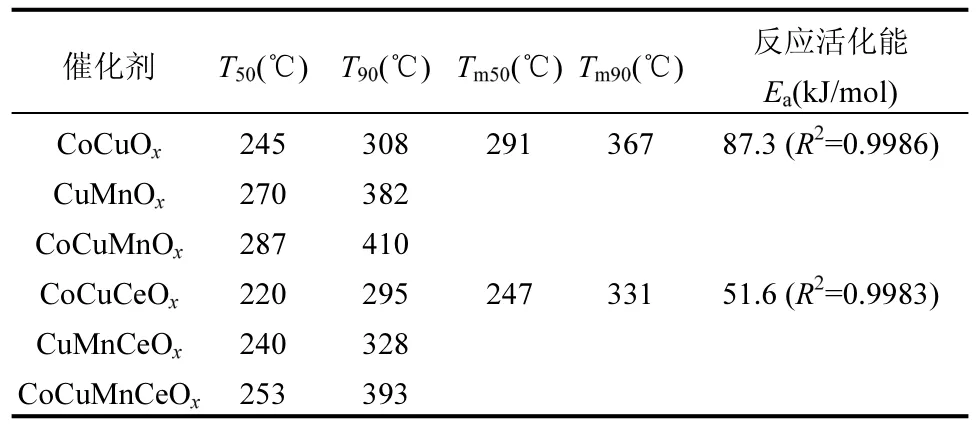
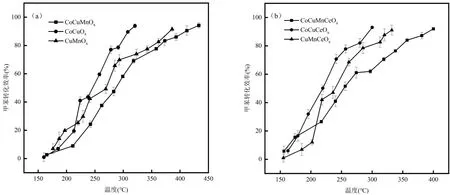
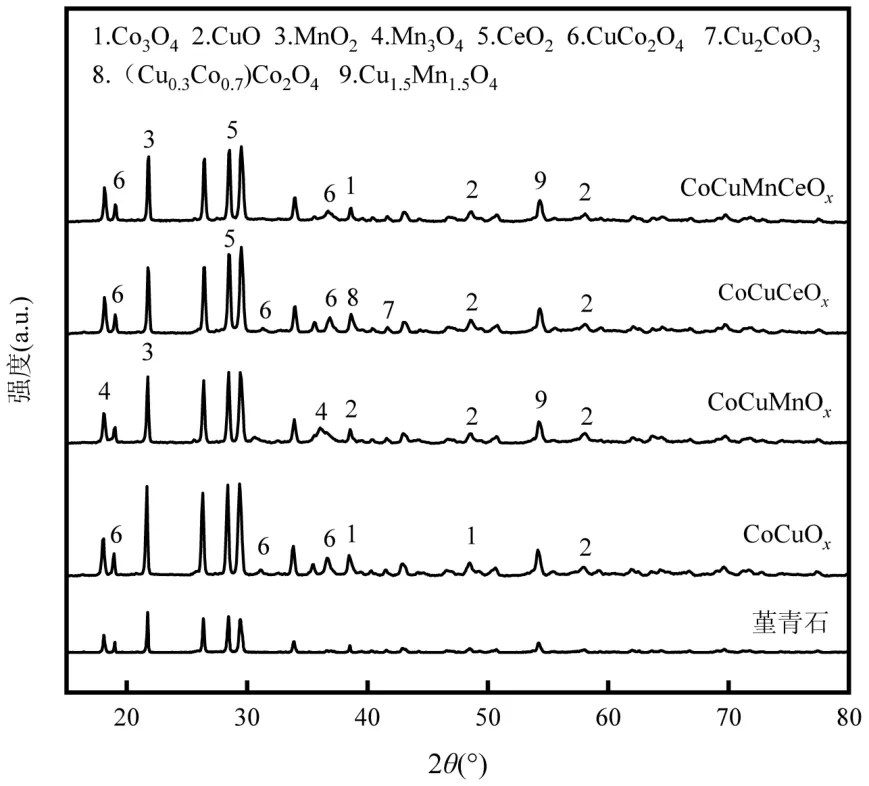
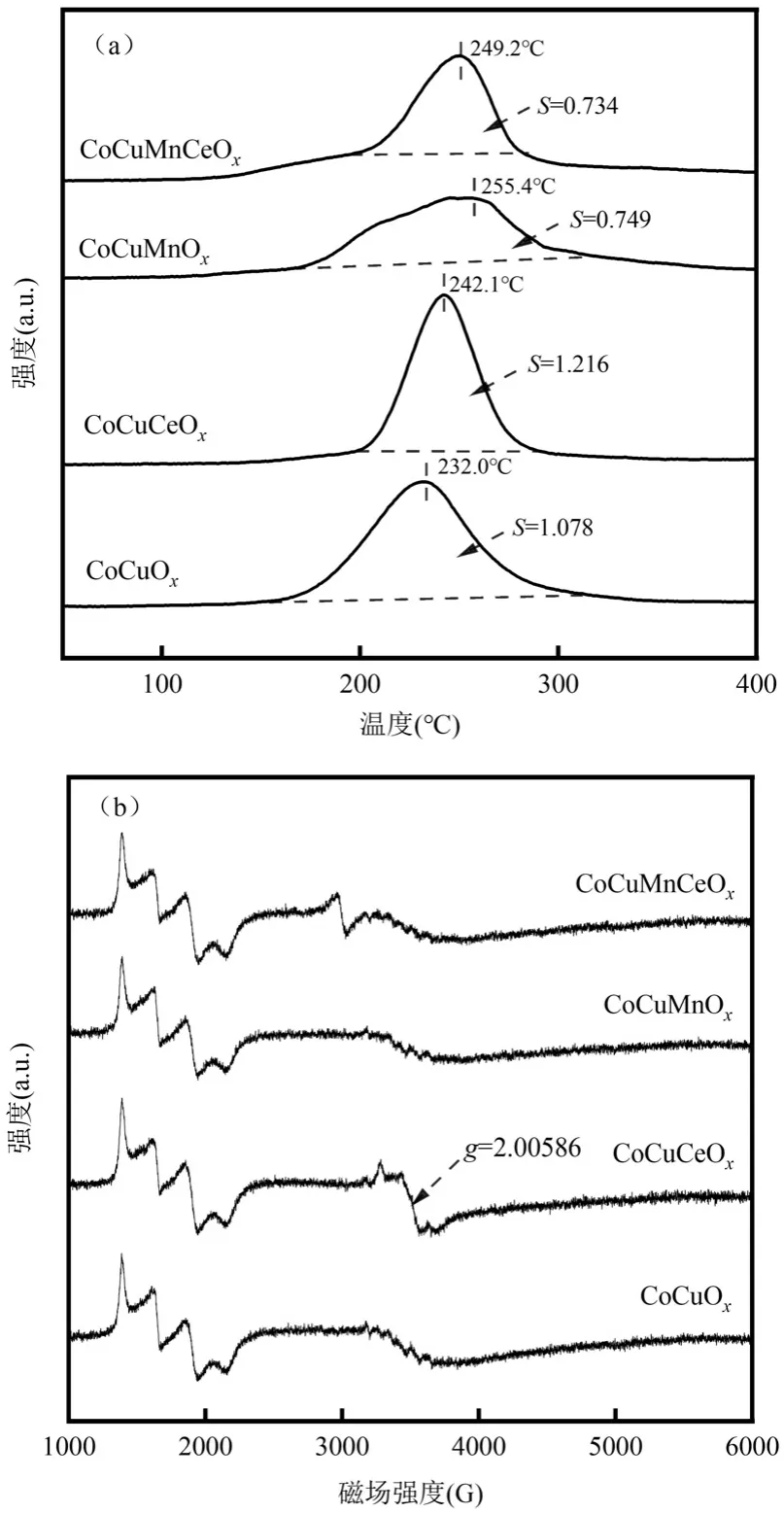
催化燃烧技术的关键是制备高效、稳定的催化剂.过渡金属Co 的高催化效率和环保性使其在多相催化领域引起了广泛关注[10].Zhang 等[11]制备了CeO2、Co3O4和CeO2-CoOx催化剂,甲苯转化率90%时,床层温度的顺序为:CeO2-CoOx(258℃) 1.1 材料与试剂 整体式蜂窝状堇青石,150mm×150mm×150mm(孔尺寸1mm×1mm),巩义市品诚环保科技有限责任公司;硝酸钴(Co(NO3)2·6H2O),中国科隆化工有限公司;硝酸铜(Cu(NO3)2·3H2O)、硝酸锰(50% Mn(NO3)2)、硝酸铈(Ce(NO3)3·6H2O)和硅溶胶等均购自湖北七八九化工有限公司;甲苯(C7H8),国药控股陕西有限责任公司.以上化学试剂均为分析纯,试验过程中均为直接使用. 1.2 催化剂制备与表征 将堇青石载体切割成直径28mm、高150mm 的圆柱体,用超纯水清洗掉载体表面浮灰后置于105℃烘箱中过夜烘干.采用超声-等体积浸渍法制备催化剂,按照Cu:Mn:Ce:Co 质量比为3:3:1:2(其中Cu 的负载量为堇青石载体质量的3.25%)分别准确称取Cu(NO3)2·3H2O、50% Mn(NO3)2、Ce(NO3)3·6H2O、Co(NO3)3·6H2O 以及硅溶胶试剂,加入超纯水,超声溶解.将载体置于浸渍液中,当浸渍液被载体全部吸收后,将其置于100℃烘箱中烘干;然后在马弗炉中450℃下煅烧5h,自然冷却后完成催化剂的制备.采用同样的方法和不同的活性组分比例,配制不同的浸渍液,制备一元、二元、三元和四元活性组分的整体式催化剂.催化剂制备过程中,加入硅溶胶可在堇青石载体表面形成硅氧烷键,从而增强活性组分与载体之间的结合强度,有效防止活性组分的脱落,其添加量为载体吸水量的3%[16]. 采用JSM-6510LV 扫描电子显微镜(SEM,日本电子)观察催化剂的表面形貌、活性组分颗粒大小及分布情况;催化剂的晶体结构通过D8ADVANCE 型X 射线衍射仪(XRD,德国布鲁克)测定;利用THERMO SCIENTIFIC ESCALAB 250Xi 型X 射线光电子能谱仪(XPS,美国赛默飞)对催化剂表面的元素种类、相对含量以及价态变化进行分析;通过Auto Chem Ⅱ 2920 型仪器对催化剂进行氢气程序升温还原(H2-TPR,美国Micromeritics)测试,分析催化剂的氧化还原性能;采用ZMX-micro6/1 型电子顺磁共振波谱仪(EPR,德国布鲁克)分析催化剂表面的氧空位. 1.3 实验方法 实验装置主要由四部分组成,分别是气体发生装置、微波装置、测温装置和尾气吸收装置,装置示意如图1 所示.实验开始前,先将催化剂放入石英管(长360mm,外径32mm,壁厚2mm)内组成固定床,再将石英管放置在微波发生装置的单模腔内,K-型铠装热电偶探针置于催化剂中间位置监测催化剂床层的实时温度,并将石英管与热电偶连接位置密封以防漏气.实验过程中,空气首先通过变色硅胶干燥柱和活性炭吸附柱除去水分和有机物,通过转子流量计控制气体流量,然后进入甲苯蒸发瓶中;甲苯液体通过微量注射泵被定量注入到蒸发瓶中蒸发,随后被空气带出在缓冲瓶内混合均匀,然后进入催化剂固定床中进行微波催化燃烧试验.此时由微波发生器产生的微波以单模传递的方式进入单模腔对催化剂进行加热,甲苯在催化剂表面与氧反应而被降解矿化,尾气经甲醇与NaOH 溶液吸收净化后排入室外. 图1 微波催化燃烧甲苯实验装置与工艺流程Fig.1 Experimental device and process flow chart of microwave catalytic combustion of toluene 利用该实验装置开展了催化剂的吸波性能和对甲苯的催化活性试验.制备的所有整体式钴基催化剂均在微波功率70W、通入空气的条件下,记录1h 内催化剂的温度变化,以此反映催化剂的吸波性能.催化剂活性试验条件为甲苯进气浓度C0为1000mg/m3,进气流速为4L/min.通过气体采样口采集反应前后的甲苯气体,经气相色谱仪(GC6890N,美国安捷伦)和傅里叶红外光谱仪(FTIR,美国赛默飞)分别测定甲苯浓度和CO2浓度,以此计算甲苯的去除率和矿化率来评价催化剂的活性.计算方法如式(1)、(2)所示.另外,为了减少试验误差,所有催化剂活性试验均重复3次,所得数据取平均值,通过计算3次重复试验结果的标准差,给出误差棒. 甲苯去除效率表达式为: 式中:C0是进口甲苯初始浓度,C 是出口甲苯浓度,mg/m3. 甲苯矿化率表达式为: 图4为面波震级的中误差比较,虚线相连的点为MS(BB)震级中误差,实线相连的点为MS震级中误差。可以看出,多数MS(BB)震级中误差比MS震级中误差小,而比 MS震级中误差大的那些点,差值基本都在0.05以内。对这些值做算术平均,得出 MS震级的平均中误差为 0.2385,MS(BB)震级的平均中误差为0.2272,后者比前者低了4.73%。 式中:C1是出口CO浓度,C2是出口CO2浓度,×10-6;C0是进口甲苯初始浓度,mg/m3;0.26 为20℃下将进口甲苯初始质量浓度换算成×10-6的系数. 2.1 钴基复合金属催化剂的吸波性能测试 由图2 可见,单金属的CoOx、CuOx、CeOx催化剂吸波性能差,1h 内稳定在35℃左右,但是单金属的MnOx则具有良好的吸波性能,1h 内稳定在270℃.多金属的CoMnOx、CoCeOx和CoMnCeOx催化剂分别升温至 47℃、32℃和 70 ℃,而 CoCuOx、CoCuMnOx、CoCuCeOx和CoCuMnCeOx催化剂分别升温至215℃、304℃、164℃和286 ℃.对比这些催化剂的升温曲线发现,虽然单金属的CoOx与CuOx吸波升温性能较差,但是Co 与Cu 复合制备的催化剂的吸波升温性能有明显的提升,可能是因为Co 与Cu 之间能够形成结构缺陷.MnOx、CoCuMnOx和CoCuMnCeOx催化剂由于Mn 以及Co、Cu 复合金属氧化物的存在表现出最好的吸波升温性能;而CoMnOx和CoMnCeOx催化剂的吸波性能较差,说明Co 与Mn 的复合不利于催化剂吸波升温. 2.2 钴基复合金属催化剂的活性研究与反应动力学分析 选择具有良好吸波性能的6 种多金属催化剂进行微波加热下甲苯的催化降解试验,试验条件为甲苯初始浓度C01000mg/m3、进气流速4L/min,各催化剂对甲苯的降解效果见图3(a、b).从图3 和表1 可以看出,催化剂对甲苯的催化活性由高到低排序为CoCuCeOx>CoCuOx>CuMnCeOx>CuMnOx>CoCuM nCeOx>CoCuMnOx.与CuMnOx和CuMnCeOx催化剂相比,使用Co 元素替代Mn 元素后的CoCuOx和CoCuCeOx催化剂对甲苯的催化活性明显提高,T90分别降低19%和10%,为308℃和295℃.但是,向CuMnOx和CuMnCeOx催化剂中加入Co 反而降低了催化剂对甲苯的催化活性,可见Co 与Mn 同时存在时对催化剂的催化活性有抑制作用.对照图2 的吸波升温曲线与图3(a、b)的催化剂活性曲线发现,CoCuMnOx和CoCuMnCeOx催化剂的吸波性能最好,但其对甲苯的催化活性却差于吸波性能稍差的CoCuOx和CoCuCeOx催化剂,说明催化剂良好的吸波性能与其催化活性并不是绝对的正相关.吸波性能是影响催化剂活性的因素之一,而判断催化剂活性的依据是VOCs 降解时的床层温度;微波照射下,吸波性能不同的催化剂床层温度的高低与微波功率不存在线性关系.选取催化活性较好的CoCuOx和CoCuCeOx催化剂,在甲苯初始浓度C01000mg/m3、进气流速4L/min 条件下,测试其对甲苯的矿化率,结果如图3(c)所示,由图3(c)可知,CoCuOx和CoCuCeOx催化剂对甲苯的矿化能力排序为CoCuCeOx>CoCuOx,与其催化活性保持一致,说明Ce 的加入可以提高CoCuOx催化剂对甲苯的催化活性和矿化能力.对比CoCuCeOx催化剂对甲苯的转化率和矿化率发现,当床层温度为295℃时,甲苯转化率达到90%,而矿化率仅为80%;矿化率相对转化率具有一定的滞后性,这主要是由于少量甲笨在反应器中未能完全转化为CO2和H2O,以中间产物的形式存在[17].在甲苯完全转化后继续升温,中间产物被氧化降解,甲苯的矿化率进一步提高. 图2 微波辐照下(P=70W)催化剂的升温曲线Fig.2 Temperature-rising curves of different catalysts under microwave irradiation(P=70W) 表1 整体式钴基复合金属催化剂的催化温度及反应活化能Table 1 The catalytic temperature and activated energy of monolithic cobalt-based composite metal catalysts 图3 整体式钴基复合金属催化剂对甲苯的催化活性及阿伦尼乌斯曲线Fig.3 The catalytic activity and Arrhenius curves of monolithic cobalt-based composite metal catalyst for toluene 为了进一步研究CoCuOx和CoCuCeOx催化剂对甲苯的催化活性,选取其微波催化燃烧甲苯的矿化率试验数据,根据相关文献[18-21],采用一级反应拟合了富氧条件下降解甲苯的阿伦尼乌斯曲线,方程如下: 2.3 钴基复合金属催化剂的物理表征 通过观察图4(a)的SEM 照片发现,CoCuOx催化剂表面颗粒态的CoOx、CuOx和CuCo2O4尖晶石相互搭建成了有空隙的球状结构.Zhang 等[22]通过溶剂热醇解法合成了纳米中空球体结构的CuCo2O4催化剂,发现这种球状结构的形貌有利于甲苯的催化氧化.CoCuOx催化剂表面的活性组分具有高的分散度,而CoCuMnOx催化剂由于MnOx的加入,催化剂表面的活性组分颗粒出现聚集现象(图4(b)),未形成完整球状结构,降低了活性组分颗粒的分散度,而催化剂表面活性组分颗粒的高度分散可以提高催化剂的活性.同样,向CoCuCeOx催化剂(图4(c))中加入MnOx使得催化剂表面的球状结构变为花瓣状(图4(d)),出现团聚现象,降低了活性组分颗粒的分散度,进而降低了催化剂的催化性能.相反,CeOx的加入使得CoCuCeOx催化剂表面的活性组分颗粒更小,相比CoCuOx催化剂有更多的活性组分颗粒搭建在一起,形成了球状形貌,载体表面的活性组分颗粒分散更加均匀,催化剂表面活性组分颗粒的高分散度有利于提高催化剂的催化性能[23]. 图4 钴基复合金属催化剂的扫描电镜图Fig.4 SEM of different cobalt-based composite metal catalysts 图5 中堇青石载体的晶相结构与标准卡片JCPDS PDF 89-1487 有着很好的对应,并且负载后的钴基催化剂均出现了归属于堇青石的特征峰.在CoCuMnOx和CoCuMnCeOx催化剂表面均检测到了单金属氧化物MnO2晶体(JCPDS PDF 39-0375)和Cu1.5Mn1.5O4尖晶石(JCPDS PDF 35-1171),而在CoCuMnOx催化剂表面还检测到了Mn3O4晶体(JCPDS PDF 18-0803).一方面,Cu1.5Mn1.5O4尖晶石的存在,可产生丰富的活性氧,从而加强活性氧的传输[24],有利于甲苯的催化氧化;另一方面,CoCuMnOx催化剂表面锰的存在形式多为晶体,使其氧化物之间未能有效分散,活性位点减少,不利于甲苯的催化降解,这与催化剂活性试验的结果相符.CoCuMnOx催化剂未检测出钴氧化物的衍射峰,从图5 中可以看出在钴氧化物的出峰位置处出现了锰氧化物的峰,推测是由于其表面形成了Mn3O4晶体影响了钴氧化物的衍射峰. 同样地,CoCuMnCeOx催化剂表面检测到了MnO2晶体和Cu1.5Mn1.5O4尖晶石,导致了其表面钴氧化物峰强度弱于CoCuCeOx催化剂.说明锰的出现对于钴氧化物晶体的形成是不利的. 图5 不同钴基催化剂的XRD 谱图Fig.5 XRD patterns of different cobalt-based catalysts 图5 分析可知,在CoCuCeOx催化剂表面检测到了最丰富的尖晶石种类,分别有CuCo2O4尖晶石(JCPDS PDF 37-0878)、Cu2CoO3尖晶石(JCPDS PDF 21-0288)和(Cu0.3Co0.7)Co2O4尖晶石(JCPDS PDF 25-0270).CuCo2O4尖晶石结构是Cu2+取代了一部分 Co3O4尖晶石结构中的 Co2+而形成的,CuCo2O4尖晶石与CuO 的协同作用会提高催化剂的催化氧化性能[21],这与催化剂活性测试结果相一致.对比CoCuCeOx与CoCuOx催化剂发现,在原本Co3O4晶体出峰的位置,检测到了(Cu0.3Co0.7)Co2O4尖晶石.推测Ce 的存在会促进Co 与Cu 形成尖晶石结构,而尖晶石中M3+表面有丰富的氧空位[25];同时Ce3+/Ce4+可以增加催化剂表面的氧空位来促进氧的存储和释放[26],从而提高催化剂的催化活性. 2.4 钴基复合金属催化剂的化学表征 由图6(a)可见,在529.4~529.7eV 处的低结合能峰归属于晶格氧(Olatt),在531.6~531.8eV 处的高结合能峰归属于表面吸附氧(Oads).Cu、Mn、Co 和Ce等的单组分或多组分催化体系的活性通常与M-O(M=Cu,Mn,Co,Ce)的断裂有关,一般而言,吸附氧更容易参与完全氧化反应,晶格氧多与选择性氧化有关[27].吸附氧能增强催化剂的氧化还原性以及氧迁移性,从而提高催化剂活性.由表2 可见,表面吸附氧的量按以下顺序排序:CoCuCeOx>CoCuOx>CoCuMnCeOx>CoCuMnOx.Zhao 等[28]制备的Co1Mn1BHNCs 催化剂具有较高的Oads/(Oads+Olatt),决定了催化剂表面丰富的氧空位,从而有利于催化剂对甲苯的低温氧化.在催化燃烧反应中,催化剂表面的吸附氧接收电子形成富电子的O2-和O-,活性氧分子极易与气相中的VOCs 发生氧化反应而生成CO2[29].CoCuOx与CoCuCeOx催化剂吸附氧的含量相差很小,因此CoCuOx与CoCuCeOx催化剂都获得了更多的氧空位,其在甲苯氧化反应循环中更容易被气态氧补充;赵洪阳等[30]研究表明,表面活性氧具有更高的迁移率,而良好的迁移率对VOCs 的氧化起到重要作用,这与本实验CoCuCeOx催化剂具有最好的催化活性结果一致. 表2 不同钴基催化剂表面元素的XPS 拟合数据占比(%)Table 2 Proportion of XPS fitting data for surface elements of different cobalt-based catalysts(%) 由图6(b)可见,两个自旋轨道双峰伴随着震荡卫星峰,表明存在Co2+和Co3+[31].779.8eV 处的Co 2p3/2和794.8eV 处的Co 2p1/2的主峰归属于Co3+,而782.3eV 处的Co 2p3/2和796.8eV 处的Co 2p1/2的峰归属于 Co2+[21].表 2 中的 XPS 分析结果显示,CoCuCeOx催化剂具有最高的表面Co3+含量,而Co3+是催化剂催化性能优异的重要因素.催化剂表面丰富的Co3+含量会产生更多的晶格缺陷,这将导致晶格表面形成更多的氧空位[30].对于 CoCuOx、CoCuCeOx和CoCuMnOx催化剂,对比Co3+的含量发现,Ce 在CoCuOx催化剂中的取代作用对催化剂表面Co3+的含量并无显著影响,相反Mn 在CoCuOx催化剂中的取代作用则显著降低了催化剂表面Co3+的含量;Ce 的加入使得Ce 与Co 之间发生了电子传递,Co2++ Ce4+↔ Co3++ Ce3+,从而促进VOCs 的氧化降解.Wang 等[32]制备CoCeOx催化剂催化氧化甲苯,表征发现Co3O4与CeO2的协同作用增强了Co3+/Co2+和Ce4+/Ce3+的电子转移,促进了催化剂表面活性氧物种的形成,增强了活性氧物种的迁移率,导致芳环的快速断裂,从而提高了催化剂的催化活性.Zeng 等[33]制备CuCo 尖晶石复合氧化物,XPS 结果表明催化剂表面Co3+是催化氧化甲苯的主要活性位点,表面更多的Co3+和吸附氧物种可以直接参与甲苯的催化氧化反应,而CuCo2O4尖晶石结构中含有大量的Cu2+-O-Co3+结构,这有利于氧物种的吸附并产生表面活性氧物种,进而提高催化剂的催化活性.分析可知,Co3+对甲苯的氧化具有促进作用,而Co2+的催化活性则相对较差. 图6(c)显示,主要包括结合能在950.6~954.3eV范围内的Cu 2p1/2自旋轨道和结合能在932.5~934.3eV 范围内的Cu 2p3/2自旋轨道;另外,在结合能为962.0eV 和942.0eV 左右有两个震荡卫星峰,它们与Cu2+的氧化态有关.在对Cu 2p3/2进一步分析可知,Cu 2p3/2轨道峰包含两种Cu 物种,分别为结合能在932.5eV 处的Cu+及在934.3eV 处的Cu2+.Cu2+对Ce3+的部分取代有利于Ce4+向Ce3+的还原,这促进了氧化还原反应的进行,进而提高催化剂的催化氧化性能[34].从表2 可以看出,在4 种Co 基催化剂中,Cu2+含量由高到低排序为:CoCuCeOx>CoCuOx>CoCuMnCeOx>CoCuMnOx催化剂.该顺序与其活性排序相同. 图6(d)显示,在641.8eV 和653.3eV 处的峰分别对应Mn 2p3/2和Mn 2p1/2.Mn 2p3/2可以被进一步分为640.0eV、641.8eV、643.8eV 3 组峰,分别归因于Mn2+、Mn3+和Mn4+;Mn 2p1/2也可以被进一步分为651.0eV、653.2eV、655.3eV 3 组峰,分别归因于Mn2+、Mn3+和Mn4+[35].表2 给出了CoMnCeOx和CoCuMnCeOx催化剂中 Mn3+的含量,可以看出CoCuMnCeOx催化剂具有较高的Mn3+含量;文献研究发现,Mn3+在催化剂中具有良好活性[20],推测这也是CoCuMnCeOx催化剂相比CoMnCeOx催化剂具有较高催化活性的原因. 图6(e)显示了催化剂Ce 3d 的8 个光谱峰,分别标记为u1、u2、u3、u1'、u2'、u3'、v1和v1'.v1和v1'分别位于885.2eV 和902.8eV,归属于Ce3+离子的特征峰,882.5eV(u1)、889.3eV(u2)、898.4eV(u3)、901.0eV(u1')、907.1eV(u2')和916.7eV(u3')处的峰归属于为Ce4+离子的特征峰,这表明Ce3+与Ce4+共存于催化剂表面,且Ce4+是Ce的主要存在形式[11].基于峰面积计算了Ce3+和Ce4+的相对比率(见表2),CoCuCeOx催化剂表面的Ce3+物种浓度最高.据报道[34,36],Ce3+的存在说明了催化剂表面氧空位出现,其浓度越高,催化剂表面的不饱和化学键越多,因此产生的氧空位就越多,从而有利于活性氧物种的形成和VOCs 的氧化降解. 采用H2-TPR 对催化剂的氧化还原性能进行分析.氧化物催化剂的活性与低温还原峰紧密相关,较低的还原温度表明催化剂具有较高的表面氧浓度和更好的氧迁移率.图 7(a)显示,CoCuOx、CoCuCeOx、CoCuMnOx和CoCuMnCeOx分别在232.0,242.1,255.4,249.2℃处具有一个强烈的还原峰,这表明4 种催化剂均具有良好的低温还原能力.单一的强还原峰表明这4 种催化剂表面的各种氧化物都完全复合,都存在表面氧物种[35].4 种催化剂的还原峰面积大小顺序为 CoCuCeOx(1.216)>CoCuOx(1.078)>CoCuMnOx(0.749) ≈CoCuMnCeOx(0.734),H2消耗量与H2-TPR 曲线的还原峰面积成正比,还原峰面积越大表明催化剂的氧化还原能力越强[37].对比CoCuOx与CoCuCeOx催化剂发现,CoCuCeOx催化剂的还原峰温度虽然高于CoCuOx催化剂,但其还原峰面积大于CoCuOx催化剂.说明随着温度的升高,CoCuOx催化剂率先开始氧化甲苯,而CoCuCeOx催化剂氧化还原能力大于CoCuOx催化剂.张悦等[38]将不同含量的CeO2掺杂到Cu-Co-O 中制备一系列不同Ce 含量的CeO2/Cu-Co-O 催化剂,结果表明适量Ce 的添加使得催化剂的还原峰温度向着更低温方向偏移,且有更大还原峰面积.本论文中Ce 的添加虽然没有使催化剂向更低温方向偏移,但是增大了还原峰面积,表明CoCuCeOx催化剂具有较好的氧化还原活性. 如图7(b)所示,有文献报道[39], g 值在2.000 左右时可以证明催化剂表面存在氧空位.CoCuCeOx催化剂在3450G 左右出现了共振信号,在g=2.00586 时出现了O2-的信号,这为催化剂表面氧空位的存在提供了直接证据.Zeng 等[40]制备CuO-CeO2催化剂,表征显示CeO2的存在有利于提高CuO 的分散度,促进CeO2上生成新的结构缺陷来吸附大量氧物种,从而促进催化剂对甲苯的催化氧化降解.对比CoCuCeOx、CoCuMnOx和CoCuOx3 种催化剂的EPR 谱图发现,Ce 的存在是造成催化剂产生氧空位的直接原因,从而有利于吸附甲苯,使其在催化剂表面发生催化氧化反应. 对比 CoCuCeOx与CoCuMnCeOx催化剂的EPR 谱图发现,Mn 的存在使得氧空位消失,推测这也是CoCuMnCeOx对甲苯的催化氧化活性差于CoCuCeOx催化剂的原因之一. 图7 不同钴基催化剂的氧化还原性能Fig.7 Redox properties of different cobalt-based catalysts(a)H2-TPR 谱图;(b)EPR 谱图 2.5 微波催化燃烧甲苯反应机理 如图8 所示,CoCuCeOx催化剂在微波照射下迅速升温,其表面形成大量的高温热点,从而降低了甲苯催化氧化活化能[13].CoCuCeOx催化剂表面存在丰富的尖晶石种类,其中CuCo2O4尖晶石与CuO 表现出协同作用,通过Cu2++ Co2+↔ Cu++ Co3+的电子转移,促进了催化剂表面氧空位的形成,提高了催化剂的催化氧化性能[21].另外,CoCuCeOx催化剂表面存在不同价态的Ce.Ce 的加入增强了催化剂表面的储氧/释氧能力,Co与Ce之间Ce3++Co3+↔Ce4++Co2+的电子转移促进了催化剂表面活性氧物种的形成,增强了有效氧物种的迁移率.因此,在甲苯氧化反应过程中,随着催化剂表面温度的升高,高价态的金属离子对甲苯的吸附能力增强,Cu+/Cu2+、Co2+/Co3+与Ce3+/Ce4+之间的电子迁移加剧,使得空气中的气态氧分子吸附到氧空位上形成Oads, Oads从金属离子表面得到电子被活化转化为Olatt[21].吸附的甲苯与Olatt在催化剂表面发生氧化反应,最终将甲苯氧化成CO2和H2O. 3.1 不同钴基复合金属氧化物催化剂对甲苯的催化活性试验结果证实,随着床层温度的升高,催化剂的氧化性能也会升高,CoCuCeOx催化剂具有最好的催化活性,其对甲苯的T50=220 ℃,T90=295℃ .催化剂吸波性能和催化活性测试可知,催化剂的吸波性能与催化活性呈不匹配关系,催化剂的催化活性受催化剂的表面形貌、氧空位、结构缺陷以及氧化还原能力等多因素共同影响. 3.2 与其它钴基催化剂相比,CoCuCeOx催化剂具有良好的氧化还原性能,在其表面形成了具有较强电子迁移能力的CuCo2O4尖晶石物种,CuCo2O4结构中的Cu2+-O-Co3+结构可以促进氧物种的吸附,产生更多的表面活性氧物种,从而提高催化剂的活性.XPS 结果表明,CoCuCeOx催化剂具有最高的Co3+和Oads含量,且Co3+是甲苯氧化的活性位点. EPR 证实CoCuCeOx催化剂表面存在氧空位,正是催化剂表面丰富的活性氧物种和钴-铜-铈之间的协同作用才使得CoCuCeOx催化剂具有更高的催化活性. 3.3 在甲苯的微波催化燃烧反应过程中,随着床层温度的升高,催化剂表面高价态的金属离子对甲苯的吸附能力显著增强,通过Cu+/Cu2+、Co2+/Co3+与Ce3+/Ce4+之间的电子转移和丰富氧空位的活化作用,吸附的甲苯与Olatt发生催化氧化反应,最终将甲苯氧化成CO2和H2O.1 材料与方法
2 结果与讨论



3 结论
