改性赤泥负载Pt 催化剂催化氧化甲苯的性能
2024-03-28方宏萍梁文俊马连刚李桂贤贵州理工学院资源与环境工程学院贵州贵阳550003北京工业大学环境与生命学部区域大气复合污染防治北京市重点实验室北京004
方宏萍,梁文俊,马 琛,马连刚,李桂贤(.贵州理工学院资源与环境工程学院,贵州 贵阳 550003;.北京工业大学环境与生命学部,区域大气复合污染防治北京市重点实验室,北京 004)
近年来,随着工业和交通运输的快速发展,甲苯、苯、丙烷等挥发性有机化合物(VOCs)的排放量显著增加.挥发性有机化合物危害人类健康,造成严重二次的空气污染[1].甲苯作为一种致癌的挥发性有机化合物,是伴随燃料过程而来的典型尾气污染物,对人类健康构成巨大威胁.同时,甲苯会加速臭氧的消耗和光化学烟雾的形成,从而破坏生态环境[2-3].到目前为止,已经开发出各种VOCs 去除方法,包括吸附[4]、热焚烧[5]、催化氧化[6]、吸收[7]、光催化[8]、生物化学[9]、非热等离子体[9]和组合系统[10-12].在上述技术中,催化燃烧法由于降解效率高、能耗低,被认为是最有效、最有前途的处理技术,已被广泛应用于挥发性有机化合物处理[13-14].
催化燃烧技术核心问题在于高效、低成本、稳定催化剂的研发,Pt 催化剂对甲苯的催化燃烧具有较高的效率,但催化活性受载体的性质、颗粒尺寸以及Pt 的负载量等因素的影响.赤泥(RM)是氧化铝产业的固体废物,是铝工业产生量最大的固体废弃物[15].我国作为氧化铝生产大国,每年排放的赤泥产生量逐年增加,达到上亿吨.但赤泥综合利用率不足4%,大部分赤泥以填埋处理方式为主,占用大量的土地,对地下水、大气和周围地表水造成环境风险问题[16].赤泥中含有大量的氧化铝、氧化铁、二氧化硅等金属氧化物[17],在催化研究方面具有独特优势[18-19].目前,赤泥作为催化剂载体和催化剂广泛应用于多种催化反应过程,如氨分解[20]、脱硝[21]、加氢裂化和加氢脱硫[22]、CH4催化氧化[23]等.赤泥催化剂中主要活性组分为Fe2O3[24],采用改性赤泥(MRM)作为载体负载Pt,可以促使贵金属Pt 与改性赤泥载体中的活性组分产生相互促进作用,提高催化效率.然而,采用改性赤泥负载Pt,并针对Pt 与改性赤泥载体之间相互作用的研究较少.
本文以甲苯为目标污染物,采用改性赤泥为催化剂载体,采取浸渍法负载贵金属Pt,探究Pt/MRM对甲苯催化燃烧的效果,对Pt/MRM 催化剂特性进行了表征,揭示贵金属Pt 与载体的相互作用,为Pt/MRM 在甲苯催化燃烧领域的应用提供参考.
1 材料与方法
1.1 催化剂制备
因赤泥本身的强碱性、复杂的物相组成,无法直接作为催化材料使用,必须进行改性处理.采用酸溶-碱沉淀法耦合焙烧进行改性,称取50g 赤泥,加入300mL 水,再加入300mL 6mol/L 的HNO3溶液,搅拌1h 后缓慢滴加氨水,调节pH 值至8,水洗多次至pH值为中性,所得滤饼放置干燥箱中干燥至恒重,然后置于马弗炉中,500℃ ,焙烧3h.所制得的样品为改性赤泥粉末.
设置3 组平行实验,取定量氯铂酸六水合物(H2PtCl6)溶于去离子水中,向该溶液中缓慢加入改性赤泥粉末作为载体,在60℃水浴下超声搅拌3h;将搅拌好的样品放入烘箱中在110℃下干燥至恒重;然后在马弗炉中加热到500 ℃,焙烧3h;冷却到室温后进行研磨、粉碎,筛分出20~40 目作为颗粒催化剂,所得的催化剂标记为xPt/MRM, x 代表铂的质量分数,为0.1%~0.5%.
1.2 催化剂表征
采用X 射线荧光分析仪(XRF)分析赤泥改性前后的成分变化,采用XRD 衍射仪(Brucker Made D8)分析样品的晶体结构;采用傅里叶变换红外光谱仪(Bruker 公司的Vertex 70 型)分析样品中的相关化学键及部分基团的信息.采用全自动比表面及孔隙度分析仪(美国麦克ASAP 2050)对催化剂进行比表面积及孔结构分析,采用程序升温还原法(Auto Chem.II 2920)测试催化剂的氧化还原特性和催化剂中氧物种的活性.采用原位漫反射红外光谱技术(in-situ DRIFT)分析反应物在催化剂表面的吸附、氧化的物种变化.
1.3 催化剂活性评价
采用固定床反应器在常压下进行催化燃烧反应,测试催化剂催化甲苯活性.催化装置主要包括气体发生装置及固定床反应器,具体流程见图1.
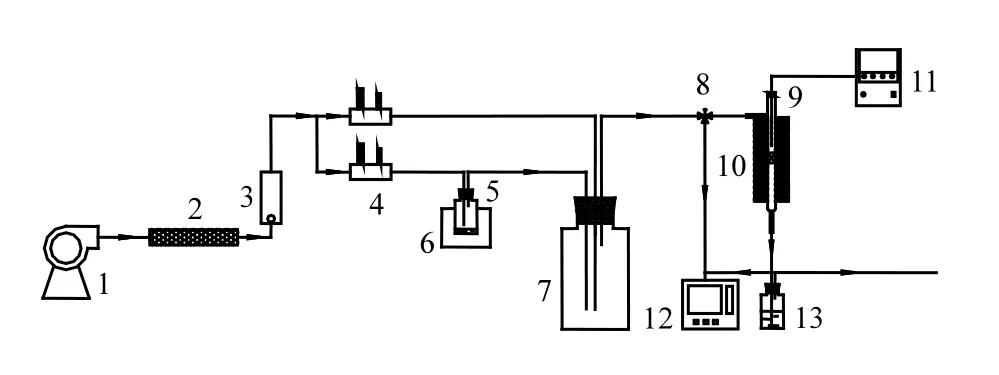
图1 催化剂活性评价流程Fig.1 Schematic diagram of catalyst activity measurement
实验条件如下:将4mL 催化剂(约2.4g)置于反应器恒温区中,催化剂床层高约15~16mm,用石英纤维棉支撑催化剂,热电偶置于催化剂床层上部,气体总气量为 2L/min,反应空速(GHSV)为 30000mL/(g·h),采用安捷伦GC7890 气相色谱对反应器出口和入口甲苯浓度进行测定.
催化剂去除效率由式(1)来表示:
式中:η 表示催化剂去除效率,%;Cin表示反应器入口气体甲苯浓度,mg/m3;Cout表示反应器出口气体甲苯浓度,mg/m3.
2 结果与讨论
2.1 Pt 含量对Pt/MRM 催化剂催化氧化甲苯活性影响
如图2 所示,原始赤泥(RM)的催化活性很差,温度为500℃时,催化效率仅为77%.Pt 直接负载在RM上,其催化活性有所提高,当温度为360℃时,0.4Pt/RM 的催化效率为100%,较原始赤泥提高了81.7%.原始赤泥因自身的强碱性和复杂的物相组成,不能直接作为催化剂,因此需要进行脱碱处理.对原始赤泥进行改性后,催化效率大幅度提升.T10(催化效率为10%时的催化温度)、T50(催化效率为50%时的催化温度)、T90(催化效率为90%时的催化温度)分别为280,325,361℃ ,温度为420℃时,催化效率为100%.改性赤泥负载Pt 后,xPt/MRM 催化剂的催化活性相较于改性赤泥均有大幅提升,Pt/MRM 催化剂的T10、T50和T90相比于MRM 显著降低,催化效率得到显著提高.MRM 催化剂在小于240℃的温度范围内甲苯的转化率为0,而负载Pt 后催化剂在180℃时就开始有甲苯被转化,在 230℃时 0.1Pt/MRM、0.15Pt/MRM、0.2Pt/MRM、0.3Pt/MRM 和0.5Pt/ MRM 的甲苯转化率分别达到11.4%、53.2%、70.5%、76.1%和 99.8%,0.4Pt/MRM 的甲苯转化率为 100%.在180~280℃的温度范围内,整体催化性能遵循以下顺序:0.4Pt/MRM>0.5Pt/MRM>0.3Pt/MRM>0.2Pt/MRM>0.15Pt/MRM>0.1Pt/MRM,0.4Pt/MRM 表现出最佳的催化活性. Zhang 等[25]采用液相氢还原方法制备了Pt1@Au1/Al2O3催化剂,Pt 的负载量为1%,甲苯进口浓度为1000×10-6,空速为18000mL/(g⋅h)时,甲苯在245℃时完全转化.Abbasi 等[26]采用湿法浸渍法制备了Pt(1wt%)/Al2O3–CeO2(30wt%)催化剂,Pt 的负载量为 1%,甲苯进口浓度为 1000×10-6,空速为5000ml/(g⋅h)时,甲苯在250℃时完全转化.本研究中制备的0.4Pt/MRM 与其他催化剂相比,具有一定的优势.

图2 Pt 含量对Pt/MRM 催化剂催化氧化甲苯活性影响Fig.2 Effect of Pt content on the catalytic activity of Pt/MRM catalyst for toluene oxidation
Pt 负载量的增加会使得催化剂表面的活性位点增多,有利于提高催化效率;而当Pt 负载量增大到一定程度时,Pt 在催化剂表面的附着以及分散度均趋于稳定,即使再增加Pt 的负载量也不能使催化剂的活性得到提高,反而略微下降,这是因为过量的Pt 物种和高负载水平下PtO 纳米颗粒的生长对甲苯的催化降解活性产生了负面影响.因此改性赤泥负载Pt 活性组分的最佳负载量为0.4wt%.
2.2 反应空速对催化剂活性的影响
从图3(a)可以看出,空速对催化氧化活性具有较大影响,在180~250 ℃,固定反应温度,随着反应空速从30000h-1升高到120000h-1, 0.4Pt/MRM 的催化效率逐步降低.这是由于空速的增加减少了污染物在反应器中的停留时间,使其与催化剂的接触时间变短,部分分子来不及被吸附于催化剂表层就从反应器离开.从另一个角度,空速增加说明单位时间内进入催化反应器内的污染物分子增加,进而导致催化剂所承担的负荷增加,从而甲苯的转化率有所下降[27].虽然反应空速提高,催化剂的甲苯催化活性下降较为明显,但当空速高达120000h-1时甲苯仍能在250℃时几乎完全被降解,说明0.4Pt/MRM 催化剂在较高空速时依然能保持着较高的催化活性.
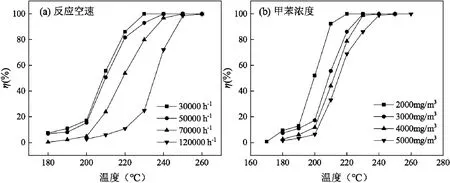
图3 反应空速和甲苯浓度对0.4Pt/MRM 催化剂活性影响Fig.3 Effect of reaction space velocity and toluene concentration on the activity of 0.4Pt/MRM catalyst
2.3 甲苯进口浓度对催化剂活性的影响
在空速为30000h-1条件下,从图3(b)可以看出,不同甲苯浓度下的甲苯转化率均随反应温度的升高而增大.但在相同的反应温度下,随着甲苯浓度从2000mg/m3升高到5000mg/m3,催化剂对甲苯的催化活性逐渐降低.在反应温度为220℃的情况下,当甲苯浓度为2000mg/m3时,甲苯已经完全转化;当甲苯浓度分别为3000,4000,5000mg/m3时,甲苯转化率分别为86.2%、78.8%和69.0%,相比于甲苯浓度为2000mg/ m3时分别降低了13.8%、21.2%和31.0%.这是由于当甲苯浓度较高时,单位时间进入催化反应器的甲苯分子数量增加,而活性位点数量固定,单位时间内催化剂处理的甲苯分子数量减少,进而降低了催化剂的催化活性.尽管甲苯浓度从2000mg/m3增加到5000mg/m3,提高了2.5 倍,但甲苯完全转化时所需要的温度分别约为220 和240℃ ,两者差值仅为20 ℃,说明0.4Pt/MRM 催化剂在较大的甲苯浓度范围内仍可以有效氧化甲苯,保持着较高的催化活性.
2.4 催化剂稳定性和重复性测试
在甲苯浓度为3000mg/m3,反应空速为30000h-1的实验条件下,从图4(a)可以看到,在214,222℃的反应温度下,随着时间的增加,催化效率分别稳定在70%和90%左右,测试过程中并无失活或恶化迹象,该催化剂具有较好的稳定性.

图4 0.4Pt/MRM 催化剂降解甲苯的反应长时间稳定性和重复使用性Fig.4 Reaction stability with time and reusability for toluene oxidation over 0.4Pt/MRM catalyst
如图4(b)所示,经过5 次循环测试后,甲苯转化率几乎没有变化,这表明该催化剂具有良好的可重复性.
2.5 催化剂表征分析
2.5.1 XRF 分析 由表1 可知,原始赤泥的各组分包括Al2O3(21%)、Fe2O3(20.4%)、CaO(17.85%)、SiO2(15.57%) 、 TiO2(4.49%) 、 Na2O(4.24%) 、MgO(2.19%)和K2O(1.04%)等.其中,Al2O3、SiO2、TiO2是常用的催化剂载体.Na2O、K2O、TiO2和MgO 为不利于催化反应发生的金属化合物,一方面,这些毒害物质会在催化过程中引起化学中毒,即占据催化剂的活性位,从而导致污染物分子无法被吸附在催化剂表面;另一方面,这些碱金属的存在不利于赤泥催化剂的制备,在煅烧环节容易致使催化剂表面烧结,造成比表面积的损失和孔结构的破坏,从而降低催化剂的活性.

表1 催化剂样品各组分质量分数(%)Table 1 Components content of the catalyst samples(%)
与RM 相比,酸改性后MRM 的各组分含量发生显著变化.MRM 的碱性物质含量相比RM 有了明显的降低.这是因为在酸洗过程中,H+除了与赤泥中NaOH、Na2CO3等可溶性碱发生反应,除去附着在赤泥表面的OH-和CO32-之外,还能通过酸解反应溶解掉Na、Ca 等碱金属物质,确保赤泥的脱碱效果.MRM 的主要成分为Al2O3、Fe2O3、SiO2和TiO2,这4 种成分总占比为73.84%,原始赤泥经改性后,成为合适的催化剂载体.
2.5.2 XRD 分析 由图5(a)可知,原始RM 是一种含多种矿物相的复合混合物,主要有方解石(CaCO3) 、 赤铁矿(α-Fe2O3) 、 钙橄榄石(Na8(Al6Si6O24)(OH)2.04(H2O)2.66)、锐钛矿(TiO2)、石英(SiO2) 、 拜三水铝石(Al(OH)3), 水钙铝榴石(Ca2.93Al1.97(Si0.64O2.56)(OH)9.44)、高岭土(Al2Si2O5(OH)4)、硅酸钙(Ca2SiO4)、硅铝酸钠(AlNa(SiO4))、硫酸钙(CaSO4),氧化铝(Al2O3).
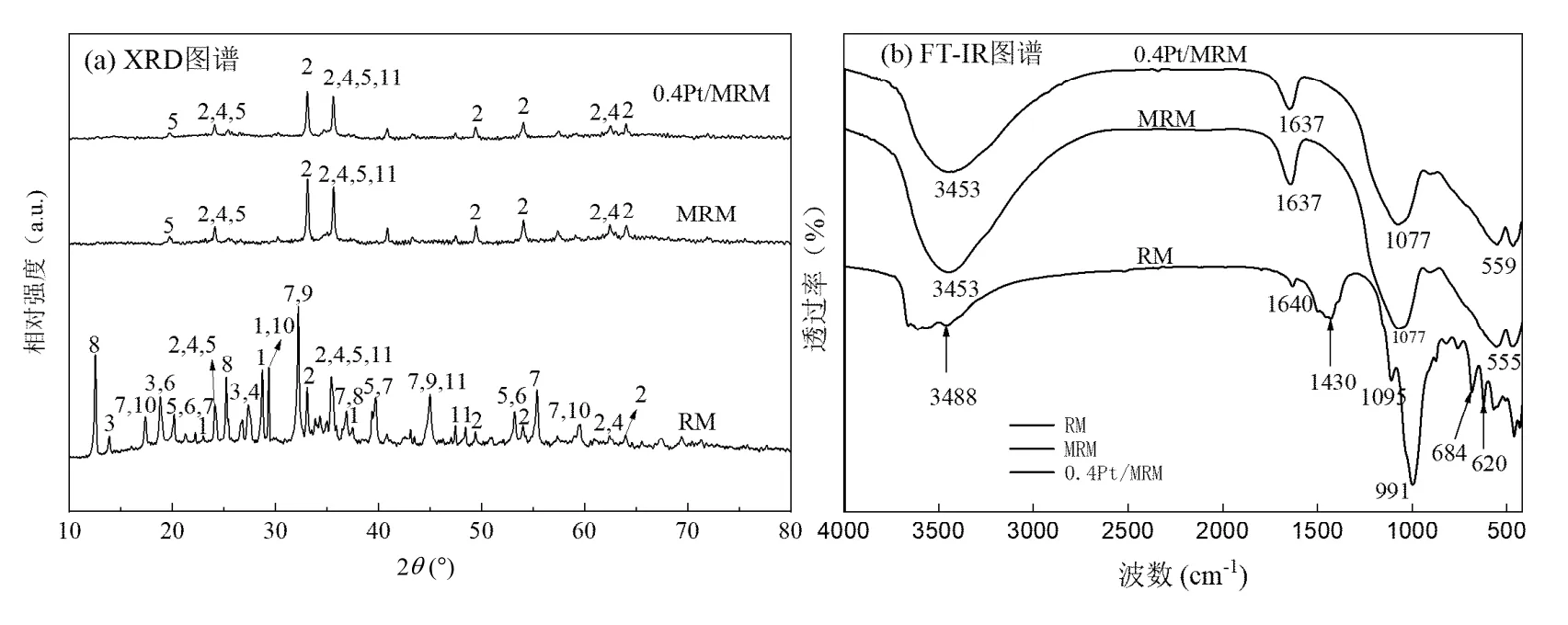
图5 RM、MRM 和0.4Pt/MRM 催化剂的XRD 和FT-IR 图谱Fig.5 XRD and FT-IR spectra of RM, MRM, and 0.4Pt/MRM catalysts
酸溶过程中赤泥含有的Na 和Ca 大部分被去除,因此可以看到MRM 归属于方解石、橄榄石、硅酸钙等的衍射峰强度相比于RM 大幅削弱.
在碱沉淀和焙烧过程中赤泥的铁含量增加,因此在24.1°、33.1°、35.6°、39.2°、49.4°、54.0°、57.5°、62.3°、63.9°处属于典型的α-Fe2O3衍射峰的峰强度相比于RM 明显提高.SiO2、Al2O3和TiO2相无明显变化,因此相关的衍射峰无明显变化.
在负载Pt 后,0.4Pt/MRM 的XRD 图谱相比于MRM 未发生明显变化,α-Fe2O3衍射峰没有消失,说明Pt 的负载没有破坏改性赤泥的晶型结构.此外,未观察到铂微晶的明显衍射峰,这可能是因为Pt 活性组分高度分散在MRM 表面上.
2.5.3 FT-IR 分析 如图 5(b)所示,在 3400,1630cm-1附近出现的峰分别由样品中水分子和赤泥铝硅酸盐结构中的H2O 分子引起[28].RM 中在1430,991cm-1处的峰说明原始赤泥含有碳酸盐成分方解石[29],而在MRM 和0.4Pt/MRM 中没有观察到,这是由于CaCO3在赤泥改性时与酸发生反应被分解.在1095cm-1和1077cm-1处的峰是Si-O 带或Al-O 带的拉伸振动,与结构Si 和Al 含量有关,684,620cm-1处的峰是Al-O 带[30],酸溶后在MRM 和0.4Pt/MRM中此峰消失.MRM 在555cm-1处的吸收峰强度明显高于RM,这归结于Fe-O 的拉伸振动.而0.4Pt/MRM中559cm-1处的吸收峰强度又略高于RM 中555cm-1处的吸收峰强度,这可能是由于Pt-O-Fe 键的拉伸振动所致[31-32].
2.5.4 BET分析 由表2可以看出,RM的比表面积为 12.9m2/g,经过改性后,MRM 的比表面积为238m2/g,提高了18 倍.比表面积的增大,使MRM 能够更好分散活性组分Pt,成为合适的活性组分Pt 的催化剂载体.
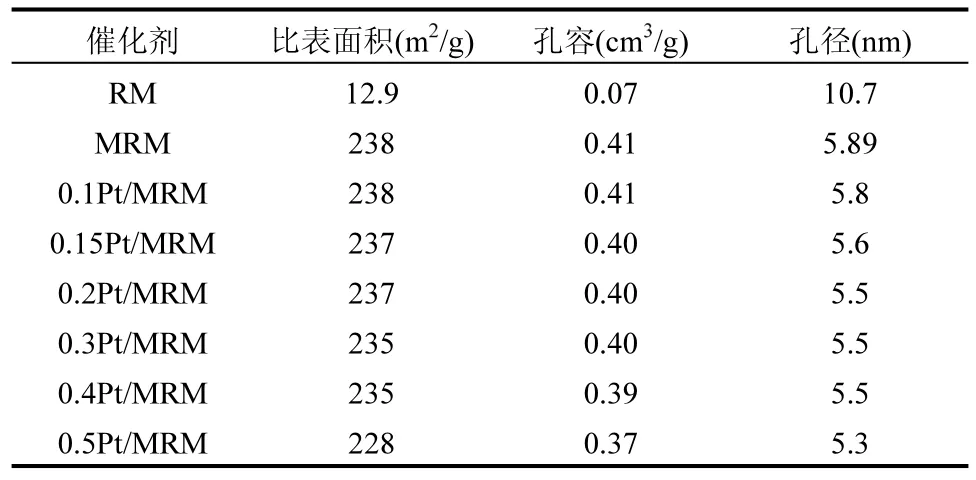
表2 催化剂比表面积及孔结构数据Table 2 Specific surface area and pore structure of catalysts
0.5Pt/MRM 的比表面积为228m2/g,相比于其他催化剂的略有减小.这可能是因为0.5Pt/MRM 催化剂的负载量过高,部分贵金属颗粒堵塞载体的孔道,且在焙烧过程中出现局部烧结[33].但是相比MRM,0.5Pt/MRM 活性仍大幅提升,这主要是因为比表面积不是决定催化剂活性的主要因素.而孔容和孔径相较于MRM 没有明显的变化,说明负载Pt 后,xPt/MRM 催化剂的孔结构未发生明显改变,高比表面积和良好的孔结构促进了甲苯分子的扩散和吸附,有利于提高其催化性能.
由图6 可知,RM 的吸脱附曲线呈现出IV 型等温线和H3 型滞后环.与MRM 相似,xPt/MRM 的吸脱附曲线均对应为IV 型等温线,滞后环在P/P0=0.4 位置处闭合,环形更靠近于H2 型滞后环,说明此催化剂属于典型的介孔材料.xPt/MRM 催化剂的吸附量没有观察到明显变化,这也说明负载Pt 后, Pt/MRM催化剂的比表面积和孔结构未发生明显改变.与其他Pt/MRM 催化剂相比,0.5Pt/MRM 的吸附量略有下降,可能是因为负载在赤泥表面的Pt 微粒出现聚集现象,导致孔道被堵塞.

图6 RM、MRM 和xPt/MRM 催化剂的N2 吸脱附曲线Fig.6 N2 adsorption and desorption curves of RM, MRM, and xPt/MRM catalysts
图7 中,峰的宽窄表明孔主要分布在对应的粒径区间内,且峰形越狭窄说明孔径趋于均一;峰值表明该孔径大小的孔数目所占比例最大.不同负载量的Pt/MRM 的孔径分布曲线与MRM 的孔径分布曲线较为接近,说明负载Pt 对催化剂的孔结构没有明显影响.xPt/MRM 催化剂依然具有大量分布在2~20nm 粒径区间的介孔,且这些介孔的粒径分布较为均一.这些介孔结构不仅增大了催化剂的比表面积,而且减小了污染物分子通过催化剂的传质阻力,有助于催化剂催化活性的提升.这与赤泥催化剂甲苯催化活性评价实验测试结果相吻合.
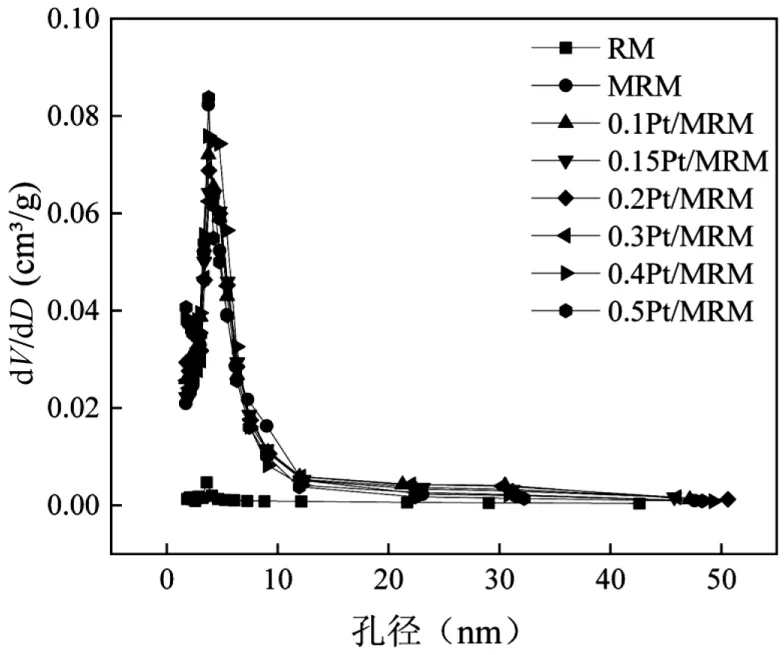
图7 RM、MRM 和xPt/MRM 催化剂的孔径分布Fig.7 Pore size distribution of RM, MRM, and xPt/MRM catalysts
2.5.5 H2-TPR 分析 如图8 所示,MRM 有2 个主还原峰(β 和γ),在300~500℃附近的峰值归因于Fe2O3→Fe3O4的还原,在500~700℃的峰值归因于Fe3O4→FeO→Fe 的还原[34].
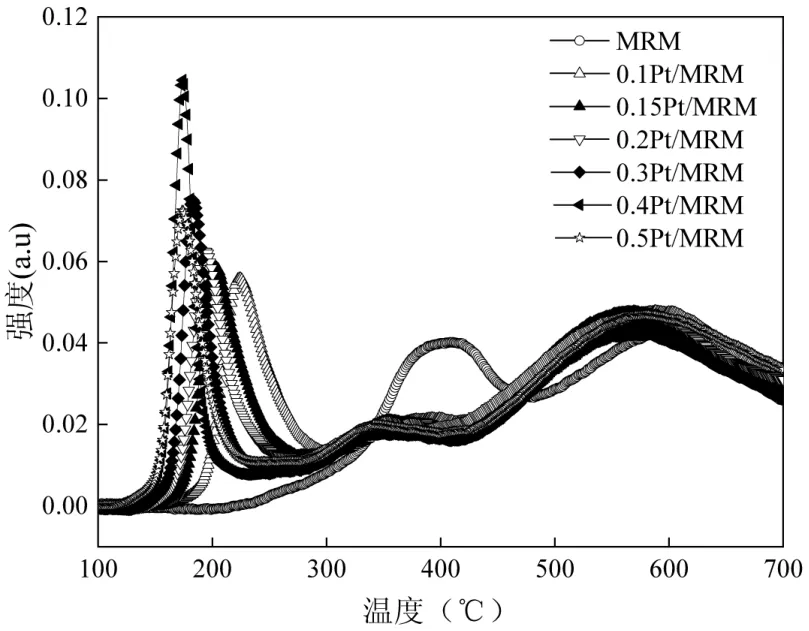
图8 MRM 和xPt/MRM 催化剂的H2-TPR 图谱Fig.8 H2-TPR spectra of MRM and xPt/MRM catalysts
不同的是,Pt/MRM 样品在100~200℃ 4 出现了一个新的宽还原峰(α),对应于H2对PtO 的还原.相比于MRM 的β 和γ 的还原峰,随着Pt 含量的增加,Pt/MRM 的还原峰(β 和γ)峰位置均向低温方向移动,表明铂与铁之间存在较强的相互作用.Pt 与Fe 间的相互作用产生的电子性质改变了催化剂的吸氢能力和双金属结构.由此可以推断,Pt 与Fe 之间的相互作用改变了Pt/MRM 催化剂中两种金属物种的还原能力,Pt-O-Fe 键的生成,有助于电子传递和氧活性物种的生成,有利于催化剂催化活性的提升[35-36].
由图9 可知,初始H2消耗速率顺序如下:0.4Pt/MRM>0.5Pt/MRM>0.3Pt/MRM>0.2Pt/MRM>0.15Pt/MRM>0.1Pt/MRM>MRM.结果表明,与MRM 相比,xPt/MRM 催化剂的催化性能大幅提升,其中0.4Pt/MRM 催化剂具有最强的低温还原能力,并且表现出最好的催化性能,特别是具有较优异的低温催化性能.这可能是因为Pt 在催化剂表面高度分散,且与Fe存在较强的相互作用.当Pt 含量增加到0.5wt%时,峰面积减小,这可能是由于Pt 在0.5Pt/MRM 催化剂中的团聚引起的,降低了催化活性.这与FT-IR 分析和催化活性测试结果相一致.
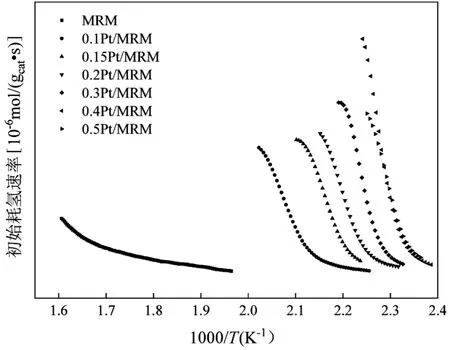
图9 MRM 和xPt/MRM 催化剂的初始H2 消耗速率曲线Fig.9 Initial H2 consumption rate of MRM and xPt/MRM catalysts
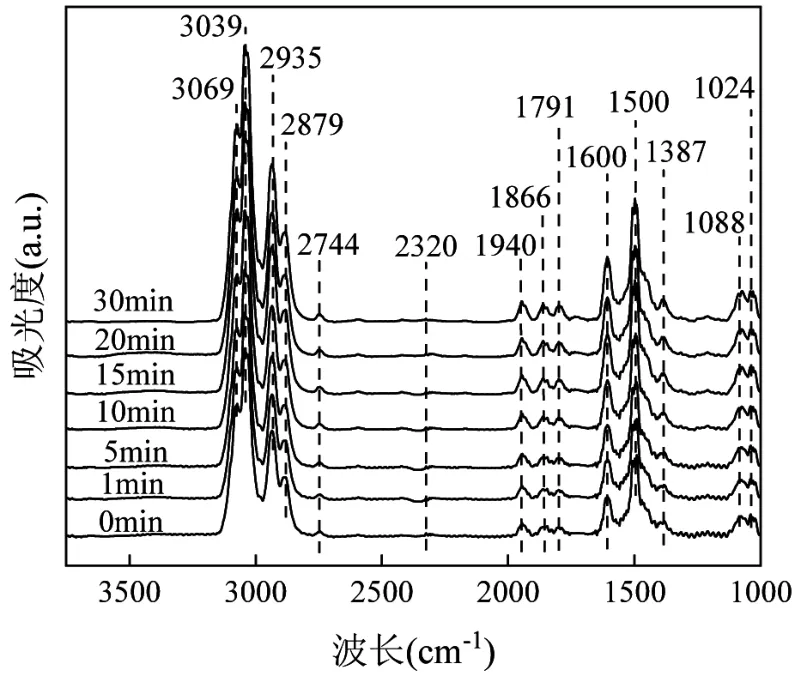
2.5.6 O2-TPD 分析 图10 显示,在100~300℃低温范围内的峰表明物理吸附的分子氧和化学吸附氧在催化剂表面上方或附近脱附,300~500℃的峰归因于催化剂中的表面晶格氧,500~800℃高温范围内的峰归因于催化剂本体晶格氧.通过计算得到100~500℃范围内的总氧解吸量由低到高的排序为RM(0.068mmol/g) 图10 RM、MRM 和0.4Pt/MRM 的O2-TPD 图谱Fig.10 O2-TPD spectra of RM, MRM, and 0.4Pt/MRM 2.5.7 反应中间产物及反应机理 图11 中,在3069和3039cm-1处的吸收峰归属为甲苯中芳香环上的C-H 键伸缩振动,在2935cm-1处的吸附峰归属为甲苯中烷基的C-H 键伸缩振动[37-38],这表明甲苯被吸收在0.4Pt/ MRM 催化剂表面.2320cm-1处的红外振动峰对应于CO2的特征振动峰[39].1387cm-1处的谱带属于醛类化合物的C=O 伸缩振动[40],表明苯甲醛的形成.在1500 和1600cm-1处出现的条带是C-O 的拉伸振动特征,是典型的羧酸基团[41-42],表明中间体中存在苯甲酸.大约在 1791cm-1、1866cm-1和1940cm-1的波段归属于五元环酸酐[42],表明有马来酸酐物种,它们是芳香环断裂过程中重要的中间体.因此,可推断甲苯燃烧的反应机理可能是: 图11 0.4Pt/MRM 催化剂在200℃条件下的表面反应原位红外光谱图Fig.11 In situ DRIFT spectra of surface reaction over 0.4Pt/MRM catalyst at 200℃ 甲苯→苯甲醇和苯甲酸→马来酸酐→二氧化碳和水 3.1 对RM 进行改性,改性后赤泥的主要成分为Al2O3、Fe2O3、SiO2和TiO2,比表面积为238m2/g,较原始赤泥提高了约18 倍,原始赤泥经改性后,成为合适的催化剂载体. 3.2 采用浸渍法制备低成本改性赤泥负载Pt 催化剂, 0.4Pt/MRM 表现出最佳的催化活性.在230℃时甲苯转化率为100%.当空速高达120000h-1时0.4Pt/MRM 催化剂依然能保持着较高的催化活性.在较大的甲苯浓度范围内仍可以有效氧化甲苯,保持着较高的催化活性. 3.3 所制备的xPt/MRM 催化剂具有较高的比表面积和良好的孔结构.在赤泥中Fe2O3与Pt 物种相互作用促进了电子转移,提高了表面氧的迁移率.Pt 物种与赤泥中Fe2O3的相互作用提高了催化效果.根据原位DRIFTS 光谱的分析,甲苯燃烧的反应机理可能是:甲苯→苯甲醇和苯甲酸→马来酸酐→二氧化碳和水.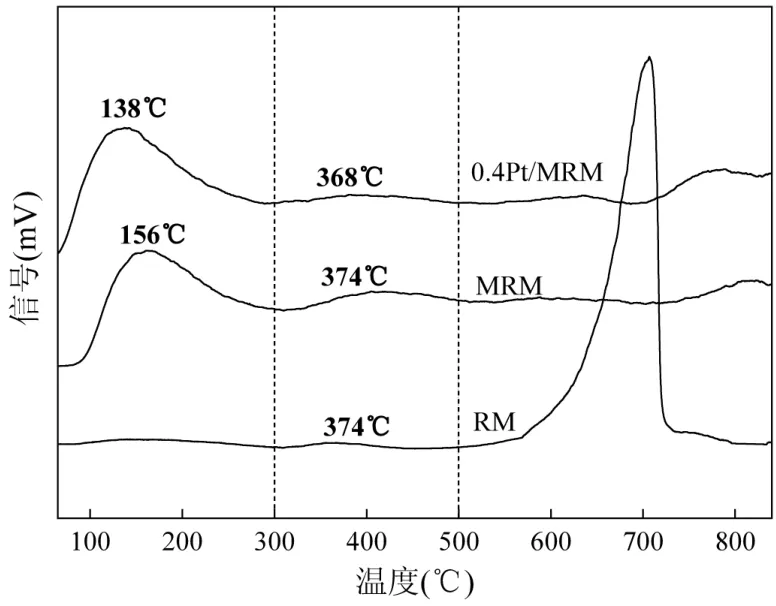
3 结论
