LZ-106诱导人肺癌NCI-H2228细胞凋亡的研究
2024-03-26蒙飞顾万建
蒙飞,顾万建
江苏省中医院 南京中医药大学附属医院 检验科,江苏 南京 210029
肺癌是当今世界上最常见的恶性肿瘤之一,也是排名第1位的肿瘤死因。非小细胞肺癌是鳞癌、腺癌和大细胞癌3种组织分型的总称,约占肺癌发生病例的85%,大多数患者确诊时已是晚期失去手术机会,整体5年生存率仅20%[1]。近年来靶向治疗因效率高、不良反应低而成为肺癌热门的研究领域及治疗方式,靶向生物标记物的开发及临床检测、靶向药物的研究和发展以及评估靶向抑制剂疗效的临床前/临床研究取得了显著进展[2]。在非吸烟肺癌中常见的驱动突变间变性淋巴瘤激酶(ALK)占非小细胞肺癌患者的5%~7%,其异常活化如基因突变、重排及扩增可激活下游的蛋白激酶B(Akt)、蛋白酪氨酸激酶(JAK)/信号转导及激活转录因子(STAT)、细胞外调节蛋白激酶(ERK)1/2等信号通路,从而导致细胞恶性转化,是非小细胞肺癌等肿瘤的致癌驱动突变基因[3]。针对该靶点的抑制剂研究已是当前抗肿瘤药物研究领域的热门方向。最先上市的是辉瑞制药公司开发的克唑替尼,在ALK阳性的晚期非小细胞肺癌治疗中取得了良好的疗效。但其严重的不良反应也逐渐引起人们的重视,如难透过血脑屏障阻碍了其在转移患者中的疗效,易原发及继发性耐药,心率不齐、脱发、胃肠不良反应及视觉障碍等严重不良反应[4]。目前,新型2、3代ALK抑制剂已取得较好的临床实验效果或已经走上临床[5-6]。此外,第4代的ALK-酪氨酸激酶抑制剂(TKI)(TPX-0131、NUV-655、Repotrectinib)也处于不同研发阶段,初步结果显示其能较好地克服复合突变,并有优越的脑渗透性。
美国食品药品管理局(FDA)于2017年批准阿来替尼为ALK阳性患者的一线用药,临床证据表明其可显著降低疾病进展或死亡风险达57%,延长患者无疾病生存时间达34.8个月,延缓脑转移的发生,给ALK阳性非小细胞肺癌患者带来明显的生存获益。阿来替尼-ALK的复合物晶体结构表明,其苯并咔唑的C-11位羰基和ALK的Met1199的氨基形成的氢键是它们相互作用的关键,此外其5位的NH和3位的氰基也与相邻Lys1150、Glu1167、Gly1269、Glu1270及Arg1253形成氢键[7]。研究表明,喹诺酮依诺沙星在多种肿瘤中具有较好的体内外抗肿瘤作用,其作用与TRBP蛋白介导的microRNA加工有关[8]。本研究通过分子对接发现在喹诺酮依诺沙星的C-3位修饰苯并咪唑的新化合物LZ-106,结构式见图1,具有相似的阿来替尼-ALK结合特征,并通过多种分子细胞生物学手段评估了其ALK激酶抑制能力、探讨了其对ALK阳性细胞凋亡的影响及机制,对于ALK抑制剂自主研发有一定参考意义。

图1 LZ-106化学结构Fig.1 Chemical structure of LZ-106
1 材料与方法
1.1 化合物及细胞系
LZ-106[2-(1-ethyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihy-dro-1,8-naphthyridin-3-yl)-1H-benzo[d]imidazole-6-carbonitrile]由中国药科大学江苏省肿瘤发生与干预重点实验室合成,为浅黄色粉末,质量分数98.5%,经DMSO配制成0.05 mol/L母液备用。阿来替尼(质量分数99.99%,货号S5232,)、Akt激活剂(SC79,质量分数97%,货号S7863)购自Selleck Chemicals公司,STAT3激活多肽(Colivelin TFA,质量分数98.86%,货号HYP1061A)购自MedChemExpress(MCE)公司。
NCI-H2228细胞系购自中国科学院上海生科院细胞资源中心,采用含10%胎牛血清[维森特生物技术(南京)有限公司,货号086-150]的RPMI-1640培养基进行培养。
1.2 分子对接
从蛋白质数据库下载ALK的蛋白结构(pdb ID:7MK7),经AutoDock程序(V 4.2)模拟ALK与LZ-106结合构象。为了尽可能完整地搜索配体的构象空间,保持蛋白质结构此期间保持固定的分子对接。进行了100次单独的遗传算法进行构象搜索,以生成100个配体的对接构象。以配体位置的质心为中心Docking box的大小为6 nm×6 nm×6 nm。Grid box设置为0.0375 nm的足够包围ALK中观察到的最大结合口袋。
1.3 分子水平激酶活性实验
根据Promega公司的ALK Kinase Enzyme System及ADP-Glo™ Kinase Assay试剂盒完成。将LZ-106、阿来替尼分别自20 nmol/L往下4倍倍比稀释10个系列梯度浓度,然后每个浓度抑制剂取1 μL加入2 ng ALK蛋白及2 μL底物室温反应60 min。加5 μL ADP-Glo™ Reagent混匀,室温反应40 min终止反应,并消耗残留ATP,然后再加10 μL激酶检测试剂室温反应30 min后经Varioskan LUX化学发光酶标仪读数。
1.4 CCK-8细胞增殖实验
将处于对数生长期的H2228细胞消化后制备成2×104个/mL的细胞悬液。设置LZ-106浓度梯度(0.25、0.5、1、2、4、8、10、12、16、24 μmol/L)并配制双倍浓度药液,将细胞悬液和药液等体积混匀,每孔液体体积为100 μL(即每孔1×104个细胞),接种于超低黏附96孔细胞培养板内,每个浓度设置6个复孔,同时设置对照(仅加细胞)和3个空白孔。药物作用24 h后,每孔加入10 μL CCK-8试剂,置于37 ℃细胞培养箱作用1~2 h后于450 nm处激发波长读取其吸光度(A)值。
存活率=(A给药-A空白)/(A对照-A空白)
1.5 Annexin V/PI细胞凋亡实验
将处于对数生长期的H2228细胞以2×105个/孔的细胞浓度,接种于6孔细胞培养板,待其贴壁并生长至适宜细胞密度(约为70%)时,设置对照组和LZ-106(1、2、4 μmol/L)组对细胞进行药物处理24 h,随后用0.25%胰蛋白酶消化并收集细胞,以PBS溶液清洗并重悬。然后依据Annexin VFITC/PI双染凋亡检测试剂盒(南京凯基生物科技发展有限公司)使用说明,用缓冲Buffer溶液重悬细胞,并将Annexin-V和PI染料分别加入样品管中,避光染色20 min后经流式细胞仪(BD FACSCalibur)检测。实验数据通过Flowjo 7.6.1软件进行分析。同法检测Akt激活剂SC79(8 μg/mL)和STAT3激活剂Colivelin(10 μmol/L)共同处理H2228细胞的诱导凋亡作用。
1.6 Western blotting法检测蛋白水平
将处于对数生长期的H2228细胞以2×105个/孔的细胞浓度接种于6孔细胞培养板,待其贴壁并生长至细胞密度约为70%时,分别加入1、2、4 μmol/L的LZ-106处理24 h。常规方法进行p-ALKY1604、p-STAT3Y705、p-ERK1/2T202/Y204、Akt、p-AktS473(Cell signaling公司),ERK1/2、Tubulin、羊抗兔二抗(ABclonal公司),ALK、STAT3(Proteintech公司)蛋白水平检测。
1.7 数据统计
2 结果
2.1 LZ-106与ALK的分子对接
应用AutoDock软件对LZ-106和ALK的分子对接构象进行模拟,结果证实了该化合物基本构型与阿来替尼基本保持一致,LZ-106化学结构图见图1。其羰基能够和ALK的关键铰链区(hinge reign)的Met1199形成氢键,7位哌嗪取代基的氨基可以与Arg1120形成氢键[7]。此外该化合物萘啶环还可以和Arg1120和Leu1122形成p-π共轭作用,这一作用是先导化合物阿来替尼所不具备的(图2)。因此推测依诺沙星衍生物LZ-106能够和ALK有效地结合并发挥作用。

图2 LZ-106和ALK模拟结构构象(A)、结构构象局部放大图(B)、LZ-106和阿来替尼在ALK蛋白中的构象叠合图(C)Fig.2 Simulated structural conformation of LZ-106 and ALK (A),local magnification of structural conformation(B),conformation superposition of LZ-106 and aletinib in ALK protein (C)
2.2 LZ-106的体外ALK激酶抑制作用
用4倍倍比(20 nmol/L起始)稀释梯度浓度的LZ-106和阿来替尼进行ALK激酶活性检测。如图3所示,二者均能浓度相关性地抑制ALK的激酶活性,半数抑制浓度(IC50)分别为(15.51±2.09)、(0.60±0.12)nmol/L,表明LZ-106能在无细胞体系中有效抑制ALK激酶活性。
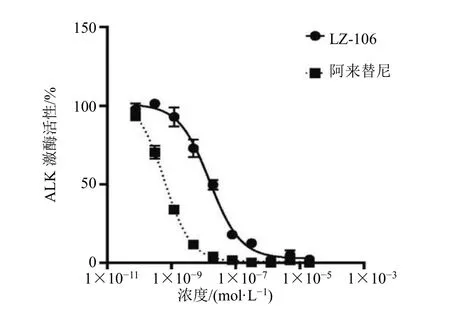
图3 LZ-106和阿来替尼对ALK激酶活性影响Fig.3 Effect of LZ-106 and alectinib on ALK kinase activity
2.3 LZ-106对细胞存活率的检测
分别用0.25、0.5、1、2、4、8、10、12、16、24 μmol/L LZ-106孵育H2228细胞12、24 h。CCK-8实验显示,不同浓度的LZ-106作用后,均表现出对H2228细胞的活力抑制作用,且其作用强度具有浓度相关性(图4),其IC50分别为(2.256±0.206)μmol/L(12 h)、(0.977±0.110)μmol/L(24 h)。

图4 LZ-106对H2228细胞存活率的影响Fig.4 Effect of LZ-106 on H2228 cell survival rate
2.4 LZ-106诱导H2228细胞凋亡
分别用0、1、2、4 μmol/L LZ-106处理H2228细胞24 h,经Annexin V/PI细胞凋亡实验检测发现,能明显浓度相关性地诱导H2228细胞凋亡(图5),其凋亡率分别为(12.78±2.56)%、(20.59±2.96)%、(37.66±4.16)%(P<0.05、0.001)。

图5 LZ-106对H2228细胞凋亡的影响Fig.5 Effect of LZ-106 on apoptosis of H2228 cells
2.5 Western blotting检测LZ-106对ALK及相关下游通路关键激酶表达的影响
如图6所示,LZ-106可以显著降低p-ALK的水平,抑制ALK的磷酸化(P<0.001)。此外研究表明,EML4-ALK通过激活下游Ras/ERK、PI3K/AKT和JAK3/STAT3通路以促进细胞恶性增殖及转移[9]。因此本研究也检测了相关通路的关键激酶表达水平,如图6所示,LZ-106处理H2228细胞后,p-ERK1/2、p-STAT3的表达水平下降(P<0.05、0.001),表明LZ-106能够浓度相关地抑制PI3K/Akt和JAK3/STAT3通路。

图6 LZ-106对H2228细胞中ALK及下游通路关键激酶活性的影响Fig.6 Effect of LZ-106 on the activity of key kinases in ALK and downstream pathways in H2228 cells
2.6 LZ-106通过PI3K/Akt和JAK3/STAT3通路诱导H2228细胞凋亡
为了进一步验证LZ-106诱导H2228细胞凋亡的作用机制,本研究采用LZ-106(4 μmol/L)与Akt激活剂SC79(8 μg/mL)、STAT3激活剂Colivelin(10 μmol/L)共同处理H2228细胞[10]。如图7所示,通过Annexin V/PI染色经流式分析可观察到SC79和Colivelin可削弱LZ-106的凋亡诱导作用,而SC79和Colivelin单独处理对细胞凋亡无明显影响。由此表明LZ-106可能通过PI3K/Akt和JAK3/STAT3通路诱导H2228细胞凋亡。
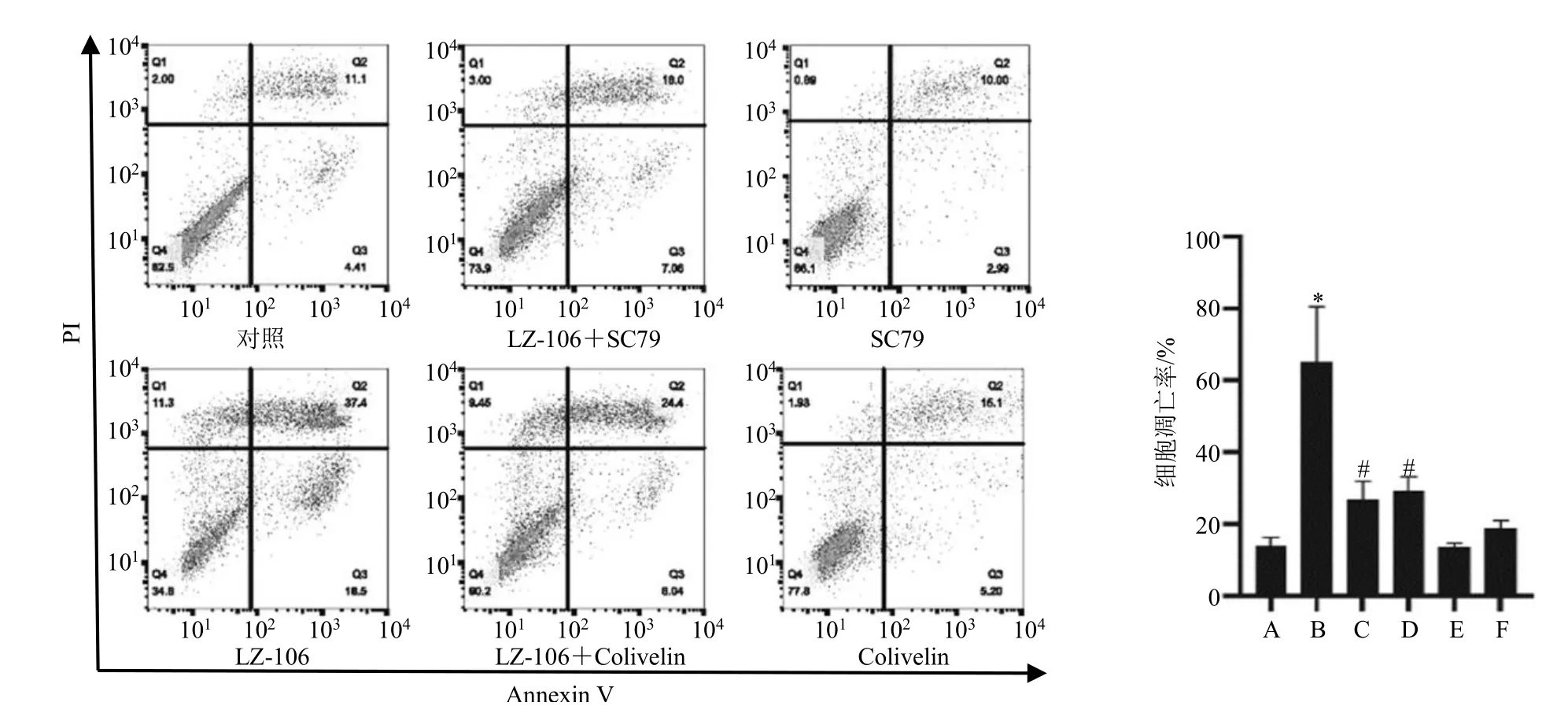
图7 LZ-106与SC79、Colivelin共同处理对H2228细胞凋亡的影响Fig.7 Effect of LZ-106 combined with SC79 and Colivelin on apoptosis of H2228 cells
3 讨论
ALK激活与数十种肿瘤的发生、发展密切相关,以ALK激酶为靶标的分子靶向药物研究逐渐成为国内外抗肿瘤药物研究的热点。LZ-106是根据阿来替尼的结构特点,在依诺沙星的结构基础上全新合成的抗肿瘤先导化合物。本研究经分子对接模拟、体外激酶实验及细胞水平等实验证实了LZ-106能显著地抑制ALK激酶活性,抑制EML4-ALK融合NSCLC的活力诱导其凋亡。且通过Western blotting以及信号通路激动剂联合处理,发现其作用机制可能是通过PI3K/Akt,JAK3/STAT3信号通路而发挥作用,但精细的作用机制还有待于进一步深入研究。
值得注意的是,本研究发现的该先导化合物虽然能与ALK结合,且具有一定的ALK激酶抑制活性,但是酶活抑制能力低于阿来替尼26倍,说明其药物结构优化与改造还有很长的路要走。此外本研究还有一些缺陷,如细胞系的选择仅仅针对1种ALK阳性细胞,后续的实验需要增加更多的细胞系;且后续需要开展异种移植瘤等动物实验进一步验证其体内的抗肿瘤效果。
综上所述,LZ-106为新一代ALK靶向药物研发提供了全新的先导化合物,能较好地抑制ALK激酶活性,促进ALK阳性细胞凋亡作用,且其对PI3K/Akt,JAK3/STAT3通路的调控可能是其作用的关键分子机制。
利益冲突所有作者均声明不存在利益冲突
