Microscopic mechanism of plasmon-mediated photocatalytic H2 splitting on Ag–Au alloy chain
2024-03-25YuhuiSong宋玉慧YiruiLu芦一瑞AxinGuo郭阿鑫YifeiCao曹逸飞JinpingLi李金萍ZhengkunFu付正坤LeiYan严蕾andZhenglongZhang张正龙
Yuhui Song(宋玉慧), Yirui Lu(芦一瑞), Axin Guo(郭阿鑫), Yifei Cao(曹逸飞), Jinping Li(李金萍),Zhengkun Fu(付正坤), Lei Yan(严蕾), and Zhenglong Zhang(张正龙)
School of Physics and Information Technology,Shaanxi Normal University,Xi’an 710119,China
Keywords: plasmon,photocatalysis,time-dependent density functional theory(TDDFT)
Plasmonic photocatalysis has made significant progress in converting solar energy into chemical energy, such as decomposition of molecules,[1]artificial photosynthesis,[2]and synthesis hydrocarbon fuels.[3]The microscopic mechanism of plasmon-induced photocatalysis has been attributed to local electromagnetic-field enhancement,[4]charge transfer,[5,6]and local thermal effects[7]generated by plasmon relaxation.Among them, transfer of hot carriers plays a crucial role in plasmon catalytic reactions.[8,9]Hot elctrons generated by plasmon decay can be transferred to reactant molecules through direct electron transfer or indirect electron transfer process.[10,11]In the direct electron transfer process, surface hybrid states can be formed because of strong interaction between the adsorbate and the metal, and plasmonic coupling with the surface hybrid states can generate hot electrons on the reactant directly.[12]In the indirect electron transfer process,the excited hot electrons interact with surrounding electrons and then transfer to the reactant molecules due to the electron tunneling effect.[8]
Alloy nanostructures with unique plasmonic property can significantly enhance the chemical reaction and is widely used in the plasmonic photocatalysis.[13]Ag-Au alloys have unique LSPR,[14]tunable optical properties,[15]and good chemical resistance,[16]usually better than pure metal nanostructures on photocatalysis.[17]Yueet al.[18]have found that the photocatalytic H2-evolution rate has been increased with a factor of eight by transferring Ag nanoparticles into Ag-Au alloy nanoparticles due to the plasmon-induced stronger electromagnetic field.Liet al.[19]have discovered that Au@Ag nanoalloys exhibit about 1.6 times higher molecular decomposition efficiency compared to Au or Ag, and proposed the enhanced photocatalytic activity owing to more hot carriers on bimetallic nanoparticles by plasmon decay.In this sense,it is still disputed which kind of microscopic mechanisms is the most effective for alloy plasmon-enhanced reaction.
Here, we investigate the real-time dynamics of H2photosplitting on alloy Ag-Au chains irradiated by femtosecond laser pulses to reveal the atomic-scale reaction mechanism by TDDFT.[20,21]The splitting rate of H2is found to depend on the position and proportion of Au atoms in alloy chains, and specially designed Ag-Au alloy is more likely to induce H2photosplitting than pure Ag chain.Moreover the difference of H2photosplitting rate can be explained by comparing the occupation evolutions of the anti-bond states of H2adsorbed on different alloy chains.This research provides a deep understanding of the atomic-scale mechanism of plasmon-mediated photochemistry on the alloy nanostructures.
Electronic structures and dynamics coupled with the ionic motion are treated within the Ehrenfest scheme.[22]The electronic structures follow the time-dependent Kohn-Sham(TDKS)equations[23]
where the functionsφiandhksare the KS orbitals and KS Hamiltonian, respectively.Table S1 presents the eigenvalue of KS orbitals for H2on Ag5-Au.The ionic motion follows the classical Newtonian equations[24]
whereMI,RI, andZIare theI-st nucleus mass, spatial location, and number of valence electrons, respectively.TDDFT can be effective in conjunction with LDA for dissociations[25]but it certainly has its limitations.Because the LDA holds that the exchange-correlation energy functional is only related to the value of the electron density at various points in space.During the process of dissociation,the change of electron density can result in the potential energy curve predicted by LDA deviating from the actual dissociation pathway.Moreover,the dissociation potential energy of LDA decreases exponentially as the relative coordinates approach infinity.Therefore,LDA is suitable for systems exhibiting a gradual change in charge density and systems with high charge density.[26]
We first compare the H2photosplitting on linear Ag5-Au and Ag6atomic chains under an external laser pulse.In our time evolution simulation of the bond length,Ag and Au atoms are considered as mobile with initial interatomic distance of 2.89 °A, chosen from the experimental value for Ag chains on NiAl (110) surface.[27]The geometry configuration of H2adsorbed on the Ag5-Au chain is shown in Fig.1(a).Here,the laser pulse polarized in thezdirection along the chain in Fig.1(b)is modeled by a Gaussian wave packet expressed as
Here,the pulse widthτis 2.6 fs,and it reaches the maximum amplitudeEmax=1.5 V/°A at the timet=9.8 fs.Due to forbidding computational costs in TDDFT quantum dynamics simulations, we utilize a strong laser field to accelerate photoreactions to make the simulations of H2photosplitting feasible.The laser frequency is chosen as ¯hω=1.54 eV, which is the plasmonic energy of Ag5-Au chain obtained from the optical absorption spectrum in Fig.S1.The laser fluence we applied is 0.08 J/cm2,which is widely available and easily applied in typical ultrafast laser experiments.[28]In addition, laser pulse duration is too short(~10 fs)to induce significant local heating or structural damage in the Ag-Au clusters.The kinetic energy of each silver and gold atom is increasing with the time and the value is in the range of 0.1-1.3 eV at the end of simulation time oft=20 fs withEmax=1.5 V/°A(Fig.S2(a)).The bondlength of Ag5-Au decreases form 2.89 °A att=0 fs to 2.85 °A att=20 fs and that of Ag1-Ag2increases from 2.89 °A to 2.98 °A (Fig.S2(b)).We estimate the cluster would be destroyed when the simulation is continued beyond 100 fs.In addition, the plasmon mode in Fig.S3 shows that the entire charge distribution remains in dipole mode.
To investigate the photo-induced dynamics response, we calculate the time-dependent H-H bond lengthdHHunder the same laser field on Ag5-Au and Ag6chains in Fig.1(c).For the sake of simplicity in the following, we will consider the H2molecule split if the bond lengthdHHis greater than 1.5 °A.On the Ag5-Au chain, the period of H2oscillation is 10.4 fs,and the bond length keeps increasing fromdHH= 0.7 °A att=10.5 fs todHH=1.9 °A att=20 fs, indicating that the H-H bond breaks on the Ag5-Au chain.Under the same condition,no dissociation is observed up tot=20 fs on the Ag6chain, despite that H2is distorted with oscillating period of 9.5 fs and the maximum bond length reaches 1.2 °A.The result suggests that H2photosplitting on Ag5-Au alloy chain is easier than that on pure Ag6chain.
In order to uncover the underlying mechanism of higher catalytic efficiency of Ag5-Au alloy chain, we compare and analyze time-evolved occupation of anti-bonding (AB) states of H2for the two complexes, fixing all atoms at initial positions under the same laser field in Fig.2(a).AB states of H2on Ag5-Au considered here are LUMO (lowest unoccupied molecular orbital)+3 and LUMO+5, where hydrogen contributes the most to the states (Fig.2(c)).The occupation of AB states is calculated by projecting the time-dependent wavefunction onto the ground state of AB states.[29]The atomic coordinates of ground state are chosen as the geometry at the timet=0 fs and not same as snapshot of the Ehrenfest dynamics.It can be identified that the occupation amount of AB states of H2on the Ag5-Au chain is greater than that on the Ag6chain, implying that the introduction of the Au atom increases the transition possibility of electron to the AB state of H2.Thus, H2photosplitting on Ag5-Au alloy chain is easier than that on pure Ag6chain because more hot electrons transfer to the AB states of H2.

Fig.2.(a) Time-dependent occupation of AB states on the Ag5-Au and Ag6 chains with a field strength of 0.05 V/°A.(b) Time-evolved transition coefficient|CLUMO+3/5,HOMO-16|2 from the occupied state HOMO-16 to AB states LUMO+3 and LUMO+5 of H2 on the Ag5-Au chain.(c)The wavefunctions of HOMO-16 (black), LUMO+3 (red), and LUMO+5 (blue) shown in boxes.Here, the isosurface value of wavefunctions is 0.1 °A-3.HOMO:highest occupied molecular orbital,LUMO:lowest unoccupied molecular orbital.
To describe the ultrafast carrier dynamics of plasmoninduced H2photosplitting on Ag5-Au alloy chain, we calculate the time-dependent transition coefficients from the occupied state (HOMO-16) showing the largest transition to AB states of H2(LUMO+3 and LUMO+5) in Fig.2(b).The wavefunctions of HOMO-16 mainly distributes on dorbital of alloy atom and that of LUMO+3 and LUMO+5 correspond to the AB states of hydrogen molecule(Fig.2(c)),indicating that electrons transfer from alloy chain to hydrogen molecule is direct charge transfer mechanism.[30]Furthermore, the other occupied orbitals contributing to LUMO+3 and LUMO+5 also have d-electron character(Fig.S4).These transition coefficients in Fig S4 display a gradual increase in amplitude, corresponding to hot electron generation by plasmon decay.[31]It can be concluded that plasmon-induced hydrogen splitting on Ag5-Au chain is owing to the direct electrons transfer from d-electron of alloy chain to the AB state of H2.Liet al.[19]have discovered that plasmonic Au@Ag nanoalloys in experiments exhibit about 1.6 times higher molecular decomposition efficiency compared to Au or Ag under visible light irradiation.Wuet al.[32]has confirmed experimentally direct charge transfer from the Au tip to CdSe nanorod with quantum efficiencies exceeding 24%.The experimental results are consistent with our theoretical results that the photocatalytic ability of Ag5-Au alloys to decompose hydrogen is 1.57 times higher than that of pure silver chains,owing to more electrons directly transfer from alloy chain to H2.
The splitting rates of H2absorbed on the Ag-Au alloy chain are also affected by the position of Au atom.The six configurations of H2absorbed on the different alloy chains are depicted in Fig.3(a).To investigate the photo-induced response, the time-dependent H-H bond lengthdHHis calculated for the six systems under the same field strength in Fig.3(b).At the end of the simulation timet=20 fs,dHHis 1.96 °A,1.4 °A,1.23 °A,0.98 °A,0.93 °A,and 0.93 °A and decreases in turn for Ag5-Au,Ag4-Au-Ag,Ag3-Au-Ag2,Ag2-Au-Ag3,Ag-Au-Ag4,and Au-Ag5,respectively.The ability of plasmons to induce hydrogen decomposition increases with Au atom approaching H2from the general trend.Obviously,when Au atom on the Ag-Au chain approaching H2, the rate of H2photosplitting increases.
To understand the influence of Au atom position in Ag-Au chains on H2splitting rate,time-evolved occupation of AB states of H2for the six complexes under the same field strength is analyzed.The occupation comparison of typical complexes of H2absorbed on Ag5-Au and Au-Ag5is shown in Fig.3(c).We can identify that the occupation amount of AB state of H2on Ag5-Au chain is greater than that on the Au-Ag5chain,resulting in the increased reaction rate on Ag5-Au chain.The occupation comparison of the other four complexes in Fig.S5 is also consistent with the H2reaction rate in Fig.3(b).The electronic structure of alloy chain could be influenced by substrate NiAl(110).Resonant energy of surface plasmon maybe shift with the substrate considered.The model of isolated Na atomic chain and Ag chain has been used for study of plasmon resonances.[33,34]The field enhancements(FE)at the position of H2in the different atom chains and the corresponding H-H bond lengthdHHatt=20 fs have been calculated (Fig.S6).The trend of FE is different with that of thedHH, indicating that FE is not the prominent drivers for the reaction.The results show that more electrons transfer from alloy chain to the AB state of H2with Au atom approaching H2,further enhancing the reaction.
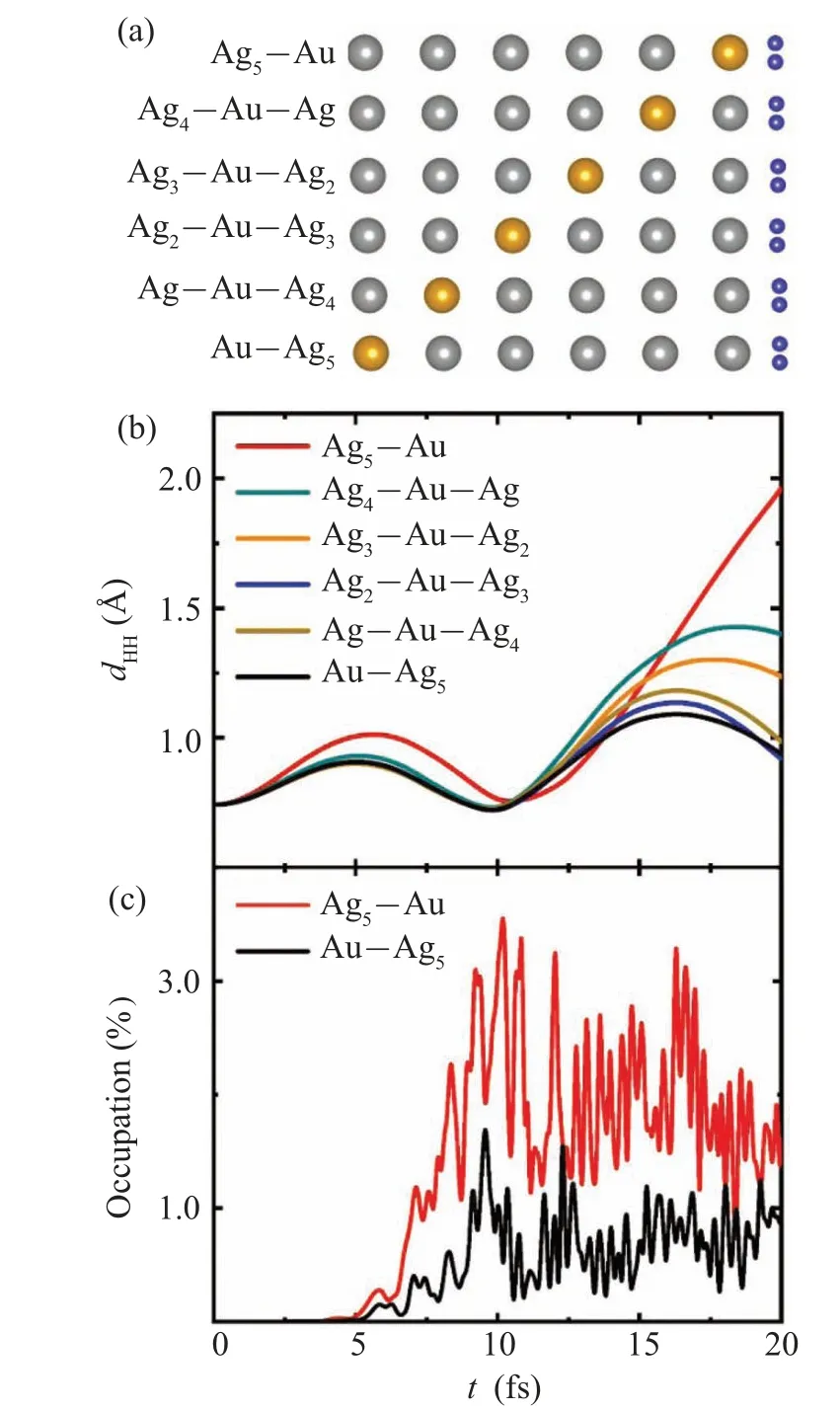
Fig.3.(a)Schematic showing H2 absorbed on Ag5-Au,Ag4-Au-Ag,Ag3-Au-Ag2, Ag2-Au-Ag3, Ag-Au-Ag4, and Au-Ag5 alloy chains.(b)Time evolution of bond length dHH under laser illumination with a maximum field strength Emax=1.5 V/°A for the six systems.Under the same laser, time-dependent occupation of AB state of H2 on Ag5-Au and Au-Ag5 chains(c).
The effect of the proportion of Au atoms in Ag-Au alloy chains on plasmon-driven reaction is further explored.The three geometric configurations of H2absorbed on Ag3-Au3,Ag4-Au2,and Au5-Ag alloy chains are depicted in Fig.4(a).To investigate the photoinduced response,the time-dependent H-H bond lengthdHHis calculated for the three systems under the same field strength in Fig.4(b).At the end of the simulation timet=20 fs,dHHis 3 °A, 2.3 °A, and 2.1 °A for Ag3-Au3, Ag4-Au2, for Au5-Ag, respectively.The H2splitting rate increases with the increased proportion of Au atoms in the Ag-Au alloy chains.In order to explain such phenomenon,time-evolved occupations of AB state of H2for the three complexes under the same field strength are analyzed in Fig.4(c).The oscillation trend of the occupations of AB states is very similar during the whole reaction process for the three complexes.The occupation amount of AB state of H2increases slightly with the increased proportion of Au atoms leading to the enhanced reaction trend in Fig.4(b)The results show that more electrons transfer from the alloy chain to the AB state of H2with increased proportion of Au atoms in the Ag-Au alloy chains,resulting in the elevated reaction rate.

Fig.4.(a) Schematic showing H2 absorbed on Ag3-Au3, Ag4-Au2,and Ag5-Au atomic chains.(b)Time evolution of bond length dHH under laser illumination with a maximum field strength Emax =1.8 V/°A for the three alloy chains.(c)Time-dependent occupation of AB states of H2 for the three complexes.
In conclusion,the plasmon-mediated H2splitting dynamics on the Ag-Au alloy chains using TDDFT is investigated.We demonstrate that the ability of plasmons to induce H2decomposition increases with Au atom approaching H2,and the increased proportion of Au atoms in the Ag-Au chains,which indicates that specially designed Ag-Au alloy is more likely to induce H2splitting than pure Ag chain.Especially,we find direct electrons transfer from the atomic chain to the AB state of H2by plasmon decay,driving the reaction.The results provide insights towards a complete fundamental understanding of plasmon-induced chemical reactions on the alloy nanoparticles at the microscopic scale and may help to improve the energy conversion efficiency in plasmon-assisted photochemistry.It is of great significance to explore the multi-scale effects of plasmon-induced chemical reaction from atomic scale to macroscopic scale.Due to substantial computational requirements of TDDFT method,calculating macroscopic cluster systems far exceeds our current computational capacity.In future work,we will extend this study to nanoscale levels such as Ag55and Ag147.
Acknowledgements
Project supported by the National Key Research and Development Program of China (Grant Nos.2020YFA0211300 and 2021YFA1201500),the National Natural Science Foundation of China (Grant Nos.U22A6005, 92150110, 12074237,and 12304426), the Natural Science Foundation of Shaanxi Province, China (Grant No.2024JC-JCQN-07), the Fundamental Science Foundation of Shaanxi Province, China(Grant No.22JSZ010),and the Fundamental Research Funds for Central Universities (Grant Nos.GK202201012 and GK202308001).
杂志排行

Chinese Physics B的其它文章
- A multilayer network diffusion-based model for reviewer recommendation
- Speed limit effect during lane change in a two-lane lattice model under V2X environment
- Dynamics of information diffusion and disease transmission in time-varying multiplex networks with asymmetric activity levels
- Modeling the performance of perovskite solar cells with inserting porous insulating alumina nanoplates
- Logical stochastic resonance in a cross-bifurcation non-smooth system
- Experimental investigation of omnidirectional multiphysics bilayer invisibility cloak with anisotropic geometry