1例Christianson综合征患儿的临床与基因变异分析
2024-03-25吕诗灵陈先睿黄建琪
吕诗灵 陈先睿 黄建琪
1.福建省厦门市康复医院儿童康复科 (福建 厦门 361003)
2.厦门大学附属第一医院儿科 (福建 厦门 361003)
Christianson 综合征(Christianson Syndrome,CS;OMIM 607379)是一种X连锁智力障碍的遗传疾病,在男性中以认知功能障碍、行为障碍和神经学表现为特征;女性携带者可表现为无症状、轻微的ID或行为问题[1]。CS临床表现为产后头小畸形、肌张力减退、吞咽困难、发育迟缓或发育倒退,重度或极重度智力障碍(Intellectual Disability,ID);行为上以孤独症谱系障碍和多动较为常见的,可有类似于天使综合症的快乐行为。由于CS患者在生命早期有口咽部吞咽困难、肌张力减退等问题,多继发出现营养不良,如体重和身高增长不良;此外还常见眼球运动异常、胃食管反流及感觉障碍,如疼痛阈值较高等[2-4]。癫痫发作通常开始于三岁之前,包括婴儿痉挛、强直、强直阵挛、肌阵挛等发作形式,因此CS患者随着年龄的增长可能失去行走和独立进食的能力。MRI常提示进行性小脑萎缩[4-5]。本研究中对1例患儿进行全外显子测序,并用Sanger测序对其双亲进行验证位点,寻找其遗传学病因以明确临床诊断,并提供遗传咨询依据。
1 临床资料
患儿男,8个月,因“生长发育落后7个月”就诊我科儿童保健门诊。患儿系G1P1,足月顺产,出生体重2.6Kg,身长49cm,无窒息抢救史。出生后发现头围小,31cm,头围增长缓慢(目前头围41cm)。1个月左右始出现肌张力偏高,无喂养困难、呕吐、反复感染等情况,抬头、握持弱,拉坐慢,可扶坐但易前倾,咿咿呀呀,声音不大。唤其名字反应 佳,可逗笑,不会翻身、独坐不能。0岁~6岁儿童发育行为评估量表示:智龄3.1个月,发育商DQ50。韦氏幼儿智力测试96分(言语理解88分,视觉空间89分,流体推理111分,工作记忆103分,加工速度89分),梦想标准化语言评估:总体语言99分(听力理解102分,语言表达89分,语义106分,句法93分)。颅脑磁共振平扫:胼胝体略纤细,双侧额颞部脑外间隙及前纵裂池增宽;双侧上颌窦及筛窦少量积液。心脏彩超:心内结构正常,左室整体缩收功能正常,舒张功能正常。脑电图:正常儿童脑电图。视听诱发电位:双耳分别刺激,中央记录:脑干听觉诱发电位:双耳Ⅰ波、Ⅲ波、Ⅴ波、各波潜伏期、各间期、波幅正常。
为明确病因,抽取先证者及其父母的外周血各5 ml(EDTA抗凝),全外显子组测序,基于高通量测序平台,采用IDT xGen Exome Research Panel进行捕获建库,双末端(Paired-End)测序策略。Raw data>10G,Q30≥80%。测序仪下机原始数据使用bcl2fastq将.bcl文件转换成.fastq文件,并使用BWA,Samtools和Picard软件将reads比对到人类参考基因组GRCh38/hg38,生成的.bam文件采用GATK系列软件进行局部重新比对,重复序列去除并进行变异检出。使用Annovar对.vcf变异文件进行变异注释。致病变异位点筛选原则:(1)筛选出外显子区变异、非同义突变位点。(2)ExAC_EAS、ExAC_ALL、1000Genomes、gnomAD等数据库中未见正常人携带或携带率小于5%。(3)参考dbSNP、OMIM、HGMD、ClinVar等多种数据库对致病变异位点进行评估。(4)使用SIFT、Polyphen2、LRT、MutationTaster、FATHMM等多种蛋白功能预测软件进行基因变异导致蛋白功能预测。根据ACMG分类指南以及病人的临床表型进行致病变异的筛选。
患儿及其双亲的高通量外显子测序数据分析发现SLC9A6基因存在c.611T>G(p.Phe204Cys)变异,父母均为野生型。根据《ACMG遗传变异分类标准与指南》6,该变异符合“临床意义未明变异”:PM6+PM2_Supporting+PP3。PM6:该变异为新发突变(不排除父母生殖腺存在嵌合可能),未经家系样本生物学关系验证;PM2_Supporting:该变异在ExAC、gnomAD、千人基因组亚洲人群数据库中的发生频率极低或没有收录;PP3:多款变异预测软件提示该变异可能有害。该基因关联疾病为X连锁遗传,患者该位点为半合子,合子类型可以解释患者患病(图1)。
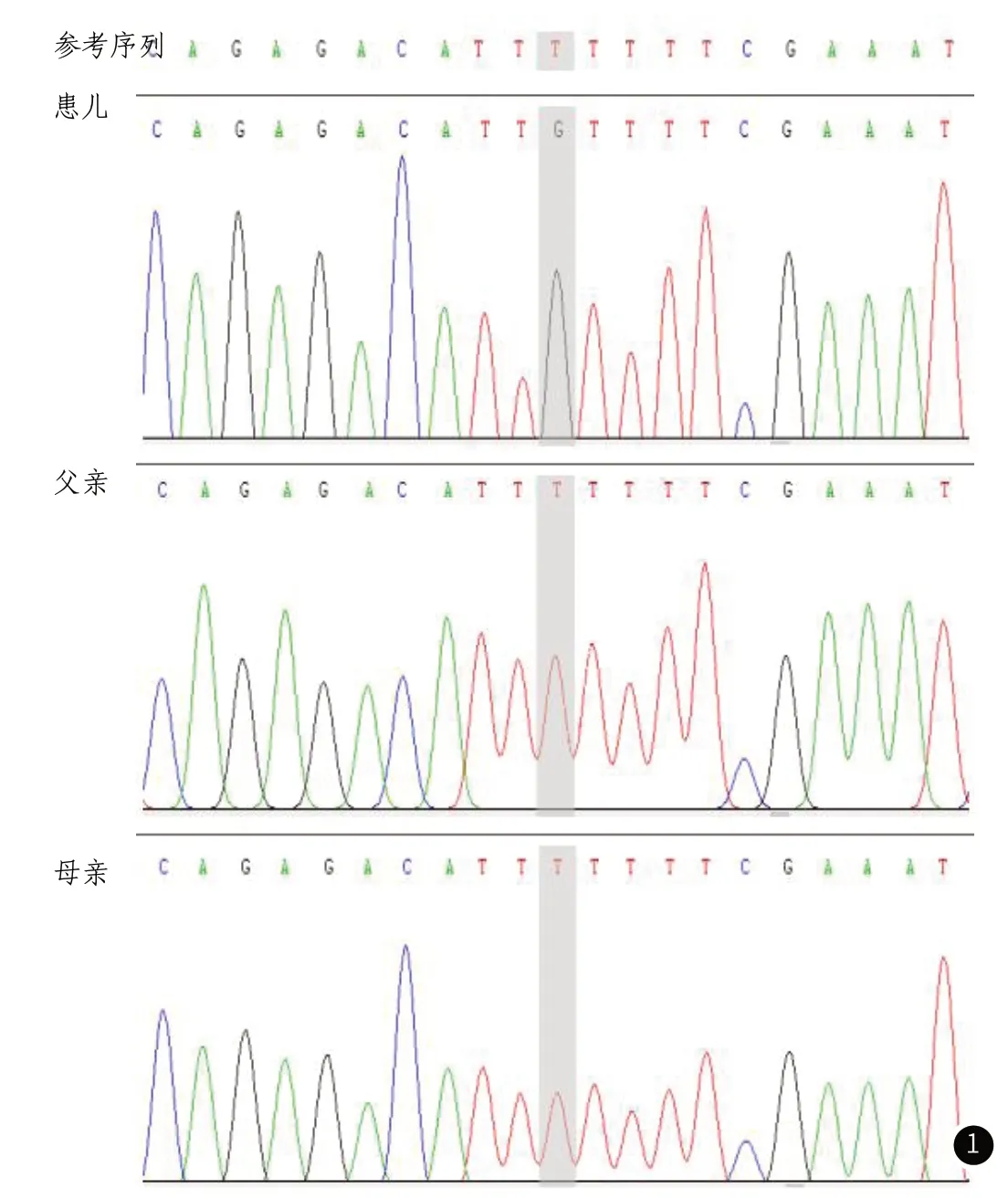
图1 先证者及家系的基因测序图。SLC9A6 :c.611T>G(p.Phe204Cys)。
2 讨 论
Christianson 综合征是一种x连锁的神经发育和退行性智力障碍疾病,根据对大约200个疑似x连锁智力残疾的家庭进行的x染色体测序,CS可能是最常见的x连锁神经发育障碍之一。男性CS的诊断是通过在X染色体上鉴定SLC9A6的半合子致病性变异而确诊,而女性携带者则需通过分子遗传识别,杂合子女性每次妊娠都有50%的机率遗传给胎儿,其中男性将会罹患CS,女性患者的表型谱广泛,可能是无症状携带者,也可能出现学习困难、语言迟缓、轻中度ID或行为问题[7-9]。男性CS患者的临床症状主要包括ID、言语受限、出生后小头畸形、多动、小脑和脑干等萎缩和神经元缺失所致的进行性躯干共济失调以及非畸形面部特征等,其他常见问题有眼动异常、感觉障碍、张力减退、胃食管反流疾病、喂养困难,尽管热量摄入正常,但始终低体重,预期寿命缩短。在临床上,CS的表型与Angelman syndrome (AS)有部分重叠,大约1/3的CS患者曾被诊断为AS[3],因此常需与AS进行鉴别诊断。其鉴别要点主要为,CS患者通常在10岁后出现进行性小脑萎缩(Cerebellar Atrophy, CA)[10],相关影像学资料提示,CS患者CA的平均发病年龄为11岁[11];另还有终身伴有体质量低下、睡眠时出现持续性癫痫状态等问题。部分人在慢波睡眠中出现癫痫状态epileptic encephalopathy with continuous spike-and-wave during slow-wave sleep (CSWS)[12],ESES常伴有睡眠诱发的癫痫放电(慢波睡眠期间连续尖峰波)和获得性认知或行为障碍,与神经认知缺陷的发展有关[13]。
SLC9A6编码蛋白属于溶质载体(SLC)超家族,是一种跨膜蛋白,编码NHE6蛋白,主要位于早期和再循环的核内体中,能够调节内体(Na+ , K+)/H+交换[5],在机体稳态条件下,通过NHE6碱化剂调节各细胞内H+-ATP酶的酸化效应。而NHE6转运体在大脑认知和智力相关功能区的发展中起着关键作用[14],广泛存在于小鼠海马神经元、树突和突触后室;在发育的神经元中,NHE6也定位于生长中的轴突束,已发现椎体神经元中NHE6的表达在突触发生过程中增加[15],总体参与神经发育、神经退行性病变和突触功能[16]。因此当SLC9A6/NHE6的功能缺失突变,导致内体-溶酶体的过度酸化、循环核内体物质的摄取受损,如转铁蛋白受体(TfR)和原霉素受体激酶B(TrkB)运输受损、上皮顶端表面极性溶解等[17-19]。核内体中的强酸性环境影响BDNF/TrkB(脑源性神经营养因子及其受体/原霉素受体激酶B)信号通路,干扰神经元生长、成熟,因此出现小头畸形、ID等表现。动物实验发现,NHE6功能敲除小鼠的视觉空间记忆和运动协调出现缺陷[20,21],而SLC9A6敲除小鼠杏仁核、海马体和大脑皮层是受影响最严重的区域[22],对热刺激和机械刺激的行为反应减少,小脑内浦肯野神经元出现退化,神经节苷质沉积[23],脊髓灰质中星形胶质细胞和小胶质细胞出现种群变化[24],提示了神经炎的发生,其结果与NHE6功能敲除小鼠表现出的神经系统症状相一致。
本例患儿运动发育迟缓、张力减退、语言缺乏、小头畸形,其X染色体测序结果提示,该患者在X染色体上的SLC9A6的第189 位氨基酸残基从苯丙氨酸Phe变成半胱氨酸Cys,第4外显子跳跃。因此,该患者被明确诊断为CS。对于CS患者的治疗,目前临床主要是支持和对症治疗,常见症状如神经发育障碍、喂养困难、癫痫发作等,临床给予相应康复治疗、抗癫痫药物等方法。由于CS早期容易误诊为AS,造成过度医疗,因此应及时对家庭成员进行分子遗传学检测。一旦在受影响的家庭成员中发现SLC9A6致病性变异,就可以对高危女性亲属进行携带者检测,对风险增加的妊娠进行产前检测,以及胚胎植入前遗传学检测。
我们的研究提供了一例由于SLC9A6基因中c.611T>G剪切位点变异引起的剪切中断,第4外显子跳跃导致蛋白质结构和功能破坏,这些分子变化进而导致了Christianson综合征的发展。对SLC9A6剪切位点变异的分子研究在临床上有助于从病理生物学和病因学上对CS进行分析,使患者得到确诊,并丰富人类基因突变数据库。
