新型冠状病毒复制子人工合成和应用研究进展
2024-03-22万里川王学军王升启
万里川,王学军,王升启
(军事医学研究院生物信息中心,北京 100850)
新型冠状病毒(SARS-CoV-2)自2019年底被发现以来[1],已在全世界范围内造成了超级大传播[2]。到2023年9月5日,新冠病毒在全球已导致7.7亿人被感染,696万人死亡(https://covid19.who.int/)。
与2002—2003年间爆发的严重急性呼吸综合征(SARS)和2012年流行的中东呼吸综合征(MERS)相似,新冠病毒感染者通常也会出现病毒性肺炎的症状,如咽喉疼痛、咳嗽、发热、疲劳、肌肉和胸部疼痛和呼吸困难等症状[3-9]。大于65岁老年人、合并有基础疾病者(如心脑血管疾病、糖尿病和肿瘤等)、免疫系统缺陷或受抑制人群、肥胖症(体质指数≥30)患者,其重症率和死亡率更高。30%左右感染新型冠状病毒的康复患者有COVID-19后遗症[10-15]。
新型冠状病毒一般通过呼吸道飞沫、密切接触或气溶胶等方式传播[16-17]。为了遏制新冠病毒的危害和传播,各国科学家研制出了不少新冠疫苗,包括传统的灭活疫苗和最新的mRNA疫苗等,对阻断新冠病毒的传播和危害起了不可或缺的作用。然而,由于新冠病毒在刺突蛋白(spike,S)关键位点的不断快速突变,使得疫苗的保护效率受到了影响[18-19]。同时,各大制药公司开发了一些针对S蛋白的鸡尾酒中和抗体和针对病毒蛋白酶的小分子化合物(如瑞德西韦和莫努匹拉韦等)对新冠患者进行治疗[20-22]。
冠状病毒科可分为α、β、γ和δ等4个属。其中属于β属的3种病毒能引发人类严重急性传染病,包括SARS-CoV-2、严重急性呼吸综合征冠状病毒(SARS-CoV)和中东呼吸综合征冠状病毒(MERS-CoV)。
冠状病毒属于单股正链RNA病毒(+ssRNA),其基因组长度为26~32 kb,是目前已知最大的RNA病毒[23]。新冠病毒基因组RNA长约29 870 bp。它和寄主mRNA结构相似,也具有5′帽子和3′多聚腺苷[ploy(A)]尾巴结构。新冠病毒基因组RNA可分为前后两个部分[24]。前面的5′端部分约占整个基因组长度的2/3,是由2个部分重叠的阅读框(ORF1a/1ab)组成,它们分别编码2个多聚蛋白pp1a和pp1ab,后者的翻译是通过在ORF1a终止密码子上游1个碱基处的程序性核糖体框架移位过程实现。这两个蛋白表达后,具有蛋白酶活性的病毒非结构蛋白NSP3和NSP5等把它们切割成16个非结构蛋白(NSPs)[25-27]。这些蛋白包括蛋白酶、RNA依赖的RNA聚合酶(RNA-dependent RNA polymerase,RdRp,NSP12)和其他装配复制-转录复合物(replication-transcription complexe,RTC)所需的蛋白,用于正义或反义的病毒基因组和亚基因组RNA合成[28]。后面的3′端部分占整个长度的1/3,编码9个附属蛋白和4个病毒结构蛋白[S蛋白、包膜蛋白(envelope,E)、膜蛋白(membrane,M)和核衣壳蛋白(nucleocapsid,N)][29-34]。
新冠病毒先是以布朗运动、扩散和静电等方式和受体细胞接触,再通过其表面的S蛋白和受体细胞膜上的血管紧张素转换酶2(ACE2)相结合。S蛋白由球状头部S1亚基和茎状S2亚基构成。S1亚基负责宿主识别,而S2亚基介导病毒和宿主细胞膜融合。与SARS-CoV的S蛋白受体结合域(RBD)相比,SARS-CoV-2 S蛋白的RBD对ACE2的结合亲和力更高。SARS-CoV-2的S蛋白上有两个切割位点,S1/S2和S2′。SARS-CoV-2的S蛋白在S1/S2位点被宿主弗林蛋白酶切割,将S蛋白转化为亚稳态。宿主细胞膜上的跨膜丝氨酸蛋白酶2(TMPRSS2)或组织蛋白酶B和L等蛋白酶在S2′位点切割S蛋白以去除S1亚基,同时激发S2亚基发生构象变化,使得病毒和宿主细胞膜融合,从而进入细胞质中[35]。
在进入受体细胞后,病毒RNA基因组首先从核衣壳结构中释放出来,利用宿主的核糖体翻译系统对病毒mRNA进行翻译,之后进行复制和转录,产生的负链模板用于基因组RNA合成,不连续合成的负链模板用于亚基因组RNA(subgenomic RNA,sgRNA)的合成,其中4个结构蛋白和9个附属蛋白均由sgRNA翻译[36]。病毒单链基因组RNA被包装在串珠型构象的N蛋白之中,形成核衣壳结构,然后以出芽的方式进入内质网-高尔基体中间体,在获得含有S、M和E等蛋白的脂质双分子层后,形成新的有感染力的病毒颗粒,再开始新一轮的生命周期[37]。
由于新冠病毒具有很强的传染性,所以有关该病毒的生物学特性、药物筛选和疫苗研发等实验工作都必须在生物安全三级及以上的实验室中展开。但是,生物安全三级及以上的实验室稀少,且实验成本高昂,实验过程烦琐,这些不利因素使得SARS-CoV-2病毒相关的研究工作受到了极大的限制[38-40]。
随着过去几十年现代病毒学的发展,科学家们发展了病毒复制子这种工具来研究高传染性病毒。所谓的病毒复制子是通过分子生物学的手段使病毒基因组中的一个或几个结构蛋白基因缺失,同时不影响病毒的复制能力。改造过的病毒复制子可以在细胞内进行正常的复制、转录和翻译,但由于缺少一个或几个结构蛋白,因而不能装配形成完整的病毒颗粒,即丧失了持续感染细胞的能力。有些病毒复制子能和结构基因(如N基因[41]、E基因或ORF3A基因)[42]或异源糖蛋白反向互补,形成有单次侵染力的病毒粒子。但在这种病毒颗粒中,仍然缺乏部分结构基因,因此在没有互补基因共转染的情况下,就只能侵染一次容纳细胞。这些复制子平台具有快速、敏感和安全的特点,是研究病毒入侵细胞和观察病毒在细胞内的复制、转录和翻译等情况以及抗病毒药物评价和筛选的理想工具[43]。
通常来说,一个典型的新冠病毒复制子包括以下四个部分[44-50]:
①启动子:如原核生物的T7启动子和真核生物的CMV启动子,分别用于在体外无细胞体系中由T7 RNA聚合酶进行转录或在体内由宿主细胞RNA聚合酶Ⅱ启动下游的病毒序列的转录。
②缺失部分结构基因的DNA或cDNA序列:通常是缺失一个或几个结构基因的部分或全长DNA或cDNA序列。
③转录终止序列poly(A):用于终止病毒序列的转录。
④报告基因:用于方便跟踪病毒在细胞体内的复制、转录和翻译等情况,通常是在结构基因如S或N基因的转录调控序列下游插入报告基因,如绿色荧光蛋白、荧光素酶或由两者组成的双报告系统[51-54]。
随着反向遗传学的发展和完善,科研人员积累了多种构建RNA病毒复制子的方法,如体外连接法、痘苗病毒载体法、BAC(bacterial artificial chromosome)载体法、酵母体内重组法(TAR)、环形聚合酶延伸反应法(circular polymerase extension reaction,CPER)等。科学家利用这些方法构建了包括微小RNA病毒科(Picornaviridae)[55],杯状病毒科(Caliciviridae)[56],黄病毒科(Flaviviridae)[57-62]和冠状病毒科(Coronaviridae)[63-67]等在内的众多的病毒复制子。其中一个有名的例子是丙型肝炎病毒的复制子,它为抗丙肝病毒感染的药物筛选提供了可靠的模型[68-70]。
常用的将复制子mRNA导入真核细胞的方法有两种:一种是利用体外无细胞转录系统,将质粒中的新冠复制子序列转录成mRNA,再通过电击或脂质体介导的方法,将mRNA导入到细胞内;第二种方法是将带有真核启动子的复制子质粒直接转染细胞,利用真核细胞本身的转录和翻译系统,把质粒里的新冠复制子序列转录成复制子mRNA序列,并翻译成相关的蛋白。第一种利用体外转录的方法,优点是可以直接得到功能性复制子mRNA,但缺点有二:一是体外转录的mRNA容易降解;二是转录过程中,如果条件控制不好,得到的全长mRNA比例会比较少,从而造成拯救效率低下。而第二种利用携带复制子的质粒来直接转染真核细胞的方法,不但转染方法比较成熟,而且mRNA表达水平通常比较高,但同一启动子在不同细胞中的启动转录的水平可能会有差异,所以对于不同种类的细胞,需要根据经验和靶细胞来决定采用合适的启动子。采用qPCR和Western blot等方法来测定病毒基因的mRNA和蛋白表达水平可以推测病毒复制子在细胞内的转录、复制和翻译情况。此外,复制子通常含有报告基因,可通过观察表达绿色荧光蛋白细胞的数量或测定荧光素酶基因表达活性等方案来间接监控病毒复制子的表达和复制情况。
新冠病毒从爆发到现在的3年多时间里,全世界科学家成功构建了多种新冠病毒不同结构蛋白缺失的复制子,进行了瞬时或稳定转染不同细胞系的研究。这些复制子为探索新冠病毒的基因功能、病毒和宿主互作机制、疫苗评价和抗病毒药物的筛选和评价等奠定了坚实的物质基础。下面就主要的四种新冠病毒复制子人工合成方法、一次性复制子和稳定表达复制子及其应用进行逐一讨论。
1 新冠病毒复制子的合成方法
1.1 通过Ⅱ型或ⅡS型限制性内切酶进行体外连接
由于新冠病毒cDNA序列长达约30 kb,加上报告基因或抗性基因就更长,很难一次从头合成全长,所以研究人员通常把设计好的全长序列分成若干段,在体外逐一化学合成或通过RT-PCR得到后,通过Ⅱ型或ⅡS型限制性内切酶酶切产生不同的黏性末端,这些黏性末端能够逐一互补配对,从而将不同片段按顺序连接起来,形成全长的病毒复制子序列。同时,复制子的首尾末端通过设计和载体的首尾端也能互补配对,从而能把全长的新冠病毒复制子序列克隆进载体。由于各个片段是分步单独克隆,很好地解决了某些病毒cDNA片段在大肠杆菌内不稳定的现象。通常在构建过程中,会删除某些病毒基因,并加上一些灵敏的报告基因,比如绿色荧光蛋白(GFP)或荧光素酶基因等,方便后续对复制子的复制和转录的观察和跟踪。根据设计原理的不同,有的复制子是通过T7 RNA聚合酶,体外转录产生病毒RNA复制子,再通过脂质体等方法转染进细胞的;而有的复制子是直接将质粒电转细胞,在细胞内转录出病毒RNA复制子。
2022年,匹兹堡大学医学院郭海涛教授团队[51]利用这种方法构建了两个目前最小的新冠病毒复制子。这两个复制子仅包含非结构蛋白ORF1a/1ab和N蛋白序列,其中一个携带T7启动子,能在体外转录成病毒复制子mRNA,而另一个是携带CMV启动子的BAC质粒。两个复制子都带有荧光素酶报告基因。构成这两个复制子的F1、F2、F3、F4这4个片段由BsaI酶切质粒得到,而F5片段和LbN片段由Esp3I酶切而来。这6个片段由于带有与上下游片段匹配的黏性末端,在T4 DNA连接酶作用下,按顺序连接得到了全长的IVT-CoV2-Rep cDNA序列,再亚克隆到可诱表达的pCC1BAC质粒中得到BAC-CoV2-Rep质粒。实验表明,由T7 RNA聚合酶体外转录得到的mRNA在转染宿主细胞后,表达的荧光素酶活性低且不稳定,可能是全长mRNA占比比较少的原因。作者考察了3种SARS-CoV-2病毒复制抑制剂——瑞德西韦和EIDD-1931(两者为RdRp酶抑制剂)、GC376(3CL蛋白酶抑制剂)对复制子的复制抑制作用。他们发现在5 μmol/L浓度时,三种抑制剂都能完全抑制复制子mRNA在CHO-K1细胞中的复制。与转染复制子mRNA相比,转染了克隆于BAC质粒中的复制子的细胞产生了强烈持久的荧光素酶活性。BAC质粒复制子转染CHO-K1细胞后,同样用5 μmol/L的这3种病毒抑制剂处理,能降低70%的N蛋白的表达,瑞德西韦只能降低约10%的荧光素酶活性,而GC376和EIDD-1931分别能降低15%和50%的荧光素酶活性。所以,他们认为,用BAC质粒复制子在细胞内产生mRNA比体外转录产生mRNA在药物筛选等实验中效果更好。总之,该研究团队成功地构建了2个迷你型的新冠病毒复制子系统,并证明可用于病毒复制、病毒寄主互作和抗病毒药物的研究中。该工作的巧妙之处在于,在这两个迷你型的复制子中,只包含了新冠病毒复制和转录必需的复制转录酶和必要的N蛋白,而删除了绝大部分结构蛋白和非必要蛋白,一方面简化了构建过程,降低了实验难度,另一方面排除了非必要蛋白对实验的干扰,可谓一举两得。
同年,罗莎琳德富兰克林医科大学的Johnny He课题组[71]利用同样的方法构建了携带双启动子和双报告基因的SARS-CoV-2复制子。这个质粒携带了HIV长末端重复序列启动子,在转染293T和Vero E6细胞后,通过HIV Tat蛋白的协助,可以进行长片段病毒RNA的复制。同时他们还检测到两个报告基因[萤火虫荧光素酶(FLuc)和GFP]的表达。另外,该复制子携带的T7启动子可以在T7 RNA聚合酶作用下,体外转录产生长的病毒RNA。经电击后,也可在Vero E6和293T细胞中复制,并检测到双报告基因的表达。药物筛选实验表明,10 μmol/L瑞德西韦处理能使多个细胞系中的报告基因表达下降70%。这些实验表明,该复制子平台可用于SARS-CoV-2病毒复制研究和抗病毒药物的筛选,具有安全、方便、快速和灵敏的特点。研究巧妙地利用了HIV长末端重复序列启动子,不仅启动效率高,而且必须要在同时转染产生的HIV Tat蛋白协助下才能启动,排除了背景非特异性启动的干扰,使实验结果可靠。另外,利用双报告系统,一方面可以直观地观察转染情况,另一方面可以对转染效果、复制效率和抗病毒药物作用效果进行定量分析。
综上所述,通过体外连接方法构建新冠病毒复制子的优势是简单易行,技术成熟。所采用的质粒可为高拷贝或中拷贝,扩增和引入突变都很容易。同时这个策略也很好地规避了把新冠病毒cDNA构建进BAC载体只有有限的酶切位点的限制。通过这些实验,我们可以看出,在体外利用T7反转录系统对新冠病毒cDNA逆转录成全长RNA并加帽,这种实验策略需要很多额外的步骤和丰富的实验经验,并且反转录产生的RNA均质性不好,因此不适用于野生型和突变型病毒复制子的定量比较分析。而利用BAC载体构建的质粒是DNA,在体外非常稳定,并且不需要体外转录和加帽等复杂的实验过程,一致性好,所以更适合用于野生型和突变型病毒复制子的定量比较分析和抗病毒药物的高通量筛选工作。
1.2 基于细菌人工染色体的构建
把病毒DNA或cDNA序列克隆进高拷贝载体是研究病毒基因功能的重要技术手段。然而,由于某些病毒DNA或cDNA序列的高表达在大肠杆菌内不能稳定存在,造成了在克隆某些全长病毒基因组时困难重重。
BAC质粒pBeloBAC11的出现成功地解决了这个问题。这个质粒包括F因子、启动子、多克隆位点、氯霉素抗性基因、LoxP位点和λcos位点等元件。由于它的复制受到了大肠杆菌F因子的严格调控,使得每个细菌体内只能产生1~2个拷贝的质粒,因而保证了外源病毒序列在大肠杆菌体内的稳定存在。2000年,Enjuanes等首先应用pBeloBAC11质粒克隆了全长的猪传染性胃肠炎冠状病毒(TGEV)[72]。随后,该方法又成功地运用到人冠状病毒OC43(HCoV-OC43)、猫传染性腹膜炎病毒(FIPV)、SARS-CoV、MERS-CoV和SARS-CoV-2等不同冠状病毒全长cDNA序列的克隆构建中[73-79]。但这个载体的缺点是拷贝数过低,提取的质粒量少,容易造成大肠杆菌DNA污染。
为了解决这个问题,Wild等把oriV复制子克隆进质粒pBeloBAC11,构建了pCC1-BAC载体[80]。该载体中的oriV复制子受TrfA复制蛋白调控,后者的表达受可诱导的启动子PBAD启动的AraC蛋白调控。经诱导后,oriV复制子可使细菌中质粒pCC1-BAC的拷贝数由1个增加到100个,因此能极大地增加质粒的拷贝数,使转染细胞所需的大量质粒很容易获得。另一个常用的克隆大片段的可诱导BAC载体是Lucigen公司的pSMART BAC载体,它通过改变氯霉素抗性基因的转录方向,避免了抗性基因转录对插入DNA片段的影响。
2021年,默沙东公司的He等科学家[52]把SARS-CoV-2病毒cDNA序列分成5个片段,在体外化学合成后,用Gibson系统把它们和酶切后的细菌人工染色体载体pSMART BAC进行重组,成功得到新冠病毒的复制子。通过体外转录和电转把RNA复制子导入不同的细胞系后,利用GFP或荧光素酶活性来观察和定量分析复制和转录活性。在转染12 h后的293T和A549细胞中,除了可以观察到GFP的表达外,还可以检测到Luc/GFP sg mRNA的转录。在电击48 h后,与293T细胞相比,A549细胞中的GFP阳性细胞数下降更快,表明复制子在293T细胞中更加稳定。这个GFP表达报告系统还表明,在其他的细胞系中,如Calu-1和Huh-7.5,复制子也能进行复制。对于293T细胞,电转效率是1.3%,而对A549、Calu-1和Huh-7.5等细胞,电转效率可达到3%~4%。为了验证GFP信号确实来自于新合成的RNA,在编码RNA依赖的RNA聚合酶的NSP12中引入2个点突变;D760N和D761N。这两个突变的联合可以使RNA依赖的RNA聚合酶失去功能,导致RNA不能合成。在转染这两个突变的复制子的A549、Calu-1和Huh-7.5细胞中,都不能表达GFP。这些数据表明,该复制子报告系统的表达来自于有活性病毒复制子RNA的复制,因此该系统可用于进行SARS-CoV-2病毒复制酶抑制剂有效性的评价。总之,该论文的实验结果表明,新冠病毒复制子能在不同细胞系中复制和转录,但在转染效率和表达上有较大的差异。同时他们巧妙地利用2个点突变来失活依赖于RNA的RNA聚合酶活性,表明转染的RNA确实发挥了病毒的转录功能,从而为体外筛选抗病毒药物奠定了坚实的理论基础。
同年,上海复旦大学的Zhang Yang等研究人员[81],以BAC为载体,构建了一个SARS-CoV-2复制子,其带有T7启动子、分泌型长腹水蚤荧光素酶报告基因,且删除了S、M和E基因。他们把体外转录加帽后的复制子mRNA与N蛋白mRNA共转Huh-7、Huh-7.5、Vero和BHK-21等不同细胞系,同时以RNA依赖的RNA聚合酶的突变体(759-SAA-761)为阴性对照。结果表明,该复制子在BHK-21细胞中的复制活性比较高,而在Vero和Huh-7.5细胞中复制活性不高。药物处理实验表明,在1~100 nmol/L浓度范围内,瑞德西韦浓度越高,荧光素酶的活性就越低。用α干扰素(IFN-α)处理复制子后,其荧光素酶活性比SAA突变体的荧光素酶活性还要低[82-83]。对于转化了抗病毒锌指蛋白长异构体(long isoform of zinc-finger antiviral protein,ZAPL,锌指抗病毒蛋白能识别非自我RNA的CPG二核苷酸而起抗病毒的作用)的Huh-7细胞,复制子转化的效率是对照组的1/10,说明该复制子的复制对ZAPL的过表达很敏感。同时,在研究中也发现丙肝病毒抑制剂达卡他韦、索非布韦和甲氧基胞苷几乎不能下调荧光素酶活性或对荧光素酶活性没有影响,说明新冠复制子只对新冠病毒的抑制剂敏感。总之,该研究同时比较了RNA依赖的RNA聚合酶突变体、瑞德西韦、IFN-α、锌指抗病毒蛋白和丙肝病毒抑制剂对新冠病毒复制子复制和转录活性的影响,对于在临床上指导新冠病毒感染的治疗有重要的参考价值。
2022年,丹麦哥本哈根大学的Bukh等学者[84]把SARS-CoV-2病毒分为4个片段,采用提取临床样品中新冠病毒RNA,经反转录PCR得到;再通过无缝克隆的方法,分3步得到含全长新冠病毒的BAC质粒,并分别构建了有感染性的病毒和无感染性的复制子。其中构建的有感染性病毒序列带有NSP12基因的5个点突变(L323P,A97V,N491S,F480L和V557L),以及在ORF7a中间部分或全部缺失的位置插入GFP、Fluc或纳米荧光素酶(nLuc)基因等报告基因。在带nLuc报告基因的复制子基础上,通过缺失部分S蛋白序列或全部S、E和M蛋白序列得到2个无感染性的病毒复制子。这2个无感染性的病毒复制子,在转染细胞后,产生的nLuc信号在转染4 h后不断增加,到24~30 h到达高峰,但在72 h后几乎检测不到。取其上清感染新的Vero E6细胞,15 d内没有出现细胞病理现象,同时免疫荧光也没有检测到N蛋白的表达,表明它们不能产生有活力的病毒。另外,瑞德西韦对这2个无感染力的病毒复制子表现出浓度依赖的抑制作用,25 µmol/L的瑞德西韦能下调荧光素酶活性至低于对照组的1/100,而低浓度2.5 µmol/L的处理能使荧光素酶活性下调至低于对照组的1/5~1/10。总之,该研究团队不仅构建了缺失不同片段的新冠病毒复制子,而且还讨论了不同报告基因在不同位置的表达效果和持续时间,对后来的实验人员构建类似的质粒提供了宝贵的经验。
需要注意的是,以BAC质粒为基础的新冠病毒复制子构建通常需要把长达30 kb的基因组分成6个以上的片段,每个片段长度控制在5 kb以下,因为大于5 kb的片段不容易克隆进中间载体,并且在凝胶回收时可能会断裂。中间过渡克隆需要进行测序验证,以防PCR带来不必要的突变。相邻的片段是通过唯一的酶切位点进行连接,以保证克隆方向的正确。如果没有合适的酶切位点,可以通过引入沉默突变,产生新的酶切位点,同时该突变也可用于病毒克隆的遗传标记。然而,多次克隆和多次测序造成了步骤烦琐,过程漫长。同时,某些病毒片段在大肠杆菌体内不稳定,这些都是BAC克隆方法的缺点。为了克服这些局限性,科学家们发展了利用酵母的重组克隆系统。
1.3 利用酵母转化相关重组进行克隆
用常规质粒克隆全长病毒序列主要有两个困难:一是可插入片段大小的限制;二是某些病毒序列在大肠杆菌中不稳定。和大肠杆菌相比,酿酒酵母(Saccharomyces cerevisiae)对外源病毒序列相对不敏感,且酵母人工染色体载体(YAC)的容量也比较大,可达250 kb[85]。研究人员利用酵母在体内能重组首尾末端有相同序列的不同DNA双链片段,发展了一种转化相关重组克隆技术(transformation-associated recombination,TAR)。它能一次把十几个甚至几十个末端有部分相同序列的片段和酵母人工染色体载体首尾相连,重组成一个新的双链DNA分子。最长的重组DNA分子长度可达110 kb以上。TAR载体一般包括一个着丝粒序列和一个酵母组氨酸选择标记。TAR载体的克隆位点和病毒复制子的首尾序列有30~110 bp相同的序列,确保两者能在酵母体内重组,连接形成一个闭环的质粒。
TAR法的原理是,酵母-大肠杆菌穿梭人工染色体载体和组成病毒复制子的多个片段在合适浓度的PEG/乙酸锂溶液中,30 ℃温浴30 min后,在一定浓度DMSO处理和42 ℃热激20 min条件下,可同时被部分酵母菌摄入体内。酵母体内天然的重组系统能把载体和复制子的不同片段按顺序重组形成一个新的质粒。没有连接复制子的空载体或只有复制子片段转化的酵母由于缺乏生成组氨酸的能力,不能生长繁殖形成克隆,而连接了外源复制子的质粒转化形成的酵母,由于具备组氨酸生成能力,能够生长繁殖形成克隆。利用PCR可以鉴定阳性含复制子的酵母菌。通过电转化等手段,可以将酵母中复制子质粒转到大肠杆菌中。通过大肠杆菌大规模制备病毒复制子后,可用于细胞转染等实验。目前,TAR克隆技术已经成功地应用于克隆大的DNA病毒,如巨细胞病毒和1型单纯疱疹病毒(HSV-1)等[86-87]。TAR可以连接的片段不仅包括从临床患者样品分离的RNA经反转录PCR得到的cDNA序列,还包括病毒的酶切片段、PCR扩增片段或化学合成的DNA片段等[88-90]。
2020年,瑞士伯尔尼大学的Jores和Thiel等研究人员[91]根据2020年1月10日公布的新冠病毒基因组序列,把它分成12个首尾末端有部分相同序列的片段(图1),除了第5和第7片段是利用从一个慕尼黑患者体内分离的SARS-CoV-2病毒进行RT-PCR得到外,其他片段由化学合成得到。之后再应用酵母转化相关重组克隆技术将这12个片段和质粒载体pVC604进行重组,从而得到6个含3种不同5′端序列的完整SARS-CoV-2病毒和用GFP取代ORF7a阅读框架内第40~282 bp序列的SARS-CoV-2病毒。从得到化学合成的片段后算起,构建全长病毒序列整个过程仅耗时7 d。这充分表明应用酵母转化相关重组克隆技术可以快速应对突发疫情。采用T7 RNA聚合酶体外转录系统产生带帽的病毒RNA,与编码新冠病毒N蛋白mRNA共转染BHK-21细胞,或单独转染稳定表达新冠病毒N蛋白的BHK-21细胞。电转后的细胞铺在Vero E6细胞上,2 d后观察到了绿色荧光信号,或产生了空斑,表明构建的病毒进行了转录和翻译,同时构建的完整病毒质粒产生了有感染力的病毒。复制动力学表明,不含GFP重组病毒和野生型的病毒没有差别或稍微下降,但在ORF7a内含GFP的病毒复制子的生长和野生型的病毒相比显著下降。血清中和实验表明,用康复期患者血清倍比稀释1∶320以内,重组病毒观察不到GFP荧光,而在1∶640稀释时可以观察到微弱荧光。同时,采用半数组织培养感染剂量(TCID50)分析法对不同浓度的瑞德西韦对SARS-CoV-2-GFP的表达抑制作用进行了研究,结果表明构建的病毒能很好地应用于抗病毒药物的筛选。总之,在新冠暴发的初期,研究人员就利用了人工化学合成的方法和酵母的快速重组系统,仅7 d就迅速构建了有活性的病毒和带荧光标记的病毒,为后继的药物筛选和阻止疫情的蔓延做出了重要的贡献。
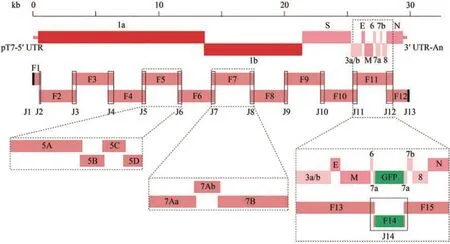
图1 基于酵母转化相关重组克隆技术的新冠病毒复制子构建[91]Fig. 1 SARS-CoV-2 genome organization and construction of SAS-CoV-2 replicon using the TAR technology[91]
2021年,洛克菲勒大学的Inna Ricardo-Lax等研究人员[43]利用酵母的反向遗传重组系统,构建了缺失了S蛋白的SARS-CoV-2复制子。他们采用Phi29聚合酶对质粒进行滚环扩增,极大地提高了质粒的产量和纯度,得到表达绿色荧光蛋白的细胞增加了20多倍。传统的复制子质粒构建时由于病毒DNA序列比较长,一般会采用低拷贝质粒,提取的质粒DNA往往量较少,该论文的科学家灵活地利用了Phi29聚合酶的高保真和高扩增的特性,为大量生产用于转染的复制子质粒提供了一种新的思路。进一步实验表明,缺失S蛋白的SARS-CoV-2复制子可以被疱疹性口腔炎病毒糖蛋白(VSV-G)包装,形成的病毒粒子能进行一次性感染,但病毒进入细胞不依赖于SARS-CoV-2的受体,如ACE2蛋白和TMPRSS2蛋白,因此极大地扩大了可感染细胞的范围。由于这种侵染只是一次性的侵染,而不是持续侵染,可以很方便地在生物安全二级实验室里进行操作。同时,他们的实验还表明该复制子系统可以用于抗病毒药物的筛选、验证和发现与新冠病毒作用的宿主因子。
综上所述,与基于BAC载体构建、体外连接等方法相比较,TAR方法具有多片段能一次性连接、效率高、无需酶切位点、连接后序列稳定和省时省力的优点。体外连接方法对连接的片段数很敏感,片段越多越不容易成功,一般都在10个以下,而TAR的方法,一次可以重组连接10个以上的片段,极大地方便了像新冠病毒这样大RNA病毒的克隆。利用BAC载体进行新冠病毒的克隆,找到合适的酶切位点往往比较困难,而TAR方法不需要酶切位点,不同片段和载体之间只需要在首尾有一段30~50 bp左右的相同序列,方便了不同突变体的构建和报告基因插入位置的选择。另外,在酵母体内,重组的全长新冠病毒序列可以快速扩繁,并且通过酵母-大肠杆菌穿梭载体,很容易把带有全长新冠病毒序列的质粒从酵母中转入大肠杆菌,方便快速产生足够量的质粒用于后续的细胞转染实验。总之,TAR方法在病毒复制子的构建特别是大病毒复制子的构建中将发挥越来越重要的作用。
1.4 应用环形聚合酶延伸反应构建
前面提到的三种方法(体外连接、基于BAC载体构建和酵母TAR系统)都要进行烦琐的病毒cDNA扩增、连接、转化或重组,以及细菌和酵母的培养和质粒的提取等步骤,费时费力。随着现代酶学技术的发展,利用PCR技术和长片段DNA聚合酶可以扩增40 kb的DNA,使得直接扩增冠状病毒基因组成为可能,无需克隆多个中间片段后连接[92-93]。2013年,Edmonds等[94]在构建黄病毒感染性克隆时,开发了这种以PCR为基础,无需借助细菌酵母的反向遗传系统,他们称之为环形聚合酶延伸反应。这种方法和前面3种方法相似的地方是,先把病毒复制子cDNA序列分为若干段,各个片段和相邻的片段之间有部分重叠的末端,再设计各个片段的特异引物,通过PCR扩出。不同的地方是,把各个片段混合退火,以自身作为引物和模板,连接产生环形的病毒复制子。这种方法的优点是产生的质粒可以直接用于转染细胞,具有快速和高保真的特点[95-97]。
2021年,日本山梨大学的Torii等研究人员[98]应用这种方法合成了新冠病毒和病毒复制子。应用这种方法,他们在2周之内就得到高滴度的新冠病毒,同时利用细菌人工染色体构建了新冠病毒复制子,它携带海肾荧光素酶和新霉素磷酸转移酶基因,瞬时转染和稳定表达该复制子的细胞株VeroE6/Rep3都能产生亚基因组RNA和病毒蛋白,并检测到荧光素酶活性。简而言之,科学家利用CPER的方法能快速构建新冠病毒的复制子,得到稳定表达复制子的细胞株,能卓有成效地进行抗新冠病毒药物筛选。
同年,澳大利亚昆士兰大学的Amarilla等科学家[99]利用CPER的方法构建了新冠病毒、木麻黄病毒、罗斯河病毒、人和鼠诺如病毒等。实验表明,构建的新冠病毒具有高度的保真性。和转染HEK293T细胞相比,转染高表达ACE2的ACE2-HEK293T细胞经过几代培养后,SARS-CoV-2病毒在弗林蛋白酶切割位点或附近产生的突变最少。细胞和动物实验表明,用CPER方法得到的病毒和父代的病毒具有相同的噬斑大小和病理变化。另外,利用CPER法还比较容易在全基因组中引入突变或插入报告基因。总之,该实验的研究人员利用CPER方法快速构建一系列活病毒和复制子,大大拓宽了CPER方法在构建不同病毒和病毒复制子方面的应用。
虽然CPER方法可以快速方便产生野生型、突变型和含有报告基因的病毒复制子,但它也有一些明显的缺点,比如:在体外PCR时,DNA聚合酶不可避免地会在病毒序列中引入预测不到的突变;同时产生环形病毒基因组cDNA的效率受设计的重叠序列、退火温度和不同循环过程中不配对等的影响较大。因此,在定量比较野生型和突变型病毒的复制方面,CPER方法不如基于BAC质粒或酵母-细菌穿梭质粒效果好。
2 单周期复制子系统
由于很多细胞在细胞膜上缺乏SARS-CoV-2病毒受体ACE2等蛋白分子,新冠病毒不容易感染成功。为了克服这个问题,新冠病毒复制子可通过和编码其他病毒糖蛋白(如水疱性口炎病毒糖蛋白)的质粒共转染,包装得到有感染性的病毒粒子,从而能入侵无ACE2受体的细胞并复制。该类型新冠病毒复制子由于缺乏感染必要的S基因,在没有继续提供外源病毒糖蛋白的情况下,仅能实现一轮感染,不能进行持续性感染,因而安全性较高,可以在生物安全二级实验室开展相关研究。
2021年,清华大学医学院丁强课题组[41]构建了用GFP取代N蛋白的新冠病毒复制子,在细胞中与同时转染并表达的SARS-CoV或SARS-CoV-2的N蛋白互补,形成有转录和复制能力的类SARSCoV-2病毒样颗粒。同时,研究者还将病毒N基因克隆到两个独立的载体,连接后的病毒N蛋白可以在病毒生命周期中发挥功能,从而进一步保证了该细胞培养模型的生物安全性。此外,他们还开发了96孔高通量抗病毒药物筛选平台,并筛选到沙林霉素、管胞苷I、莫能菌素钠、氯化霉素和尼格列宁钠等一批抗SARS-CoV-2感染的强效抗病毒药物。
同年,得克萨斯大学医学分部Shi Pei-Yong课题组[42]构建了一种反式互补系统,它能产生可单次感染的SARS-CoV-2病毒。该反式互补系统包括含有ORF3和E基因缺失的基因组cDNA以及转录调控序列突变的质粒,以及表达这两个缺失基因的细胞系。反式互补产生的病毒粒子只能感染细胞一轮,但不会产生野生型SARS-CoV-2。接种互补衍生病毒粒子的仓鼠和K18-hACE2转基因小鼠没有表现出可检测到的疾病。同时实验表明,他们构建的单周期可侵染的SARS-CoV-2复制子可应用于高通量中和检测和抗病毒药物筛选。
2022年,爱荷华大学的Malicoat等学者[100]利用BAC系统构建了一个SARS-CoV-2病毒复制子,其中S蛋白的开放阅读框架序列被长腹水蚤荧光素酶和neonGFP双报告系统取代。通过共转染该复制子BAC质粒和马水疱性口炎病毒糖蛋白表达质粒到293T/Huh-7.5的混合细胞中,拯救得到有感染性的病毒复制子颗粒,同时检测到有荧光素酶活性和观察到表达GFP的细胞。把共转染复制子和糖蛋白的Huh-7.5细胞上清转到新鲜的Huh-7.5细胞中,不能检测到荧光素酶活性和观察到GFP信号,这表明病毒复制子只能感染一次。抗VSV血清与对照血清比可以显著抑制ΔS-VRP(G)对Huh-7.5细胞的感染,证明ΔS-VRP(G)的感染是通过VSV-G蛋白的介导。在人和小鼠肺上皮细胞、肾细胞、单核细胞、淋巴细胞、巨噬细胞和树突状细胞中,ΔS-VRP(G)都能很好地侵染和复制,但表达水平不一,表明还有其他因子参与了复制过程。同时在小鼠骨髓来源的巨噬细胞和树突状细胞里,可诱导寄主强烈的抗病毒基因的表达,如α干扰素、β干扰素(IFN-β)、Mx1、Isg15和肿瘤坏死因子α(TNF-α)的表达。这些实验表明该双报告系统可以很好地应用于寄主的抗病毒反应研究。在ΔS-VRP(G)侵染Huh-7.5细胞和A549细胞后,加入不同浓度的瑞德西韦和GC376,观察到荧光素酶活性降低和GFP表达细胞减少,两者的IC50值分别为26.7 nmol/L和17.3 nmol/L。同时观察到不同的细胞对于抗病毒药物的敏感性不同。
总之,通过病毒复制子和外源的糖蛋白的互补作用,形成具有单周期感染特性的病毒,极大地扩展了病毒可侵染细胞的范围,为研究病毒基因功能、病毒和细胞的互作,以及抗病毒药物筛选和评价提供了新的思路和平台。同时,这种互补系统只能形成一次性可侵染病毒,而不能形成持续性感染的病毒,因此,该类实验可在生物安全二级的实验室里进行。最后,这种单周期可侵染病毒产生的上清可以冻存和转移,具有良好的稳定性、一致性和重复性,因此适合大规模抗病毒药物的筛选。
3 稳定表达的复制子系统
由于瞬时表达的RNA复制子对细胞有内在毒性,导致了它不能在细胞中持续地复制,造成适合复制检测的时间短,不同细胞批次之间不稳定,因而不适用于高通量筛选大化合物库。因此,构建能稳定表达SARS-CoV-2复制子的细胞株成了这个领域研究的新方向之一。
2022年,山梨大学的Kohji Moriishi课题组[101]把携带海肾荧光素酶和新霉素磷酸转移酶的融合基因的新冠病毒复制子克隆进BAC载体,通过转染细胞,建立了稳定表达新冠病毒复制子的细胞株。经检测,该细胞株能稳定表达荧光素酶,并产生病毒的亚基因组RNA。同时实验表明,用瑞德西韦和β-干扰素处理两种细胞,都能抑制病毒基因和荧光素酶的表达,而卡莫司他(一种TMPRSS2抑制剂,抑制新冠病毒进入细胞)和法匹拉韦(一种核苷的类似物,有病毒抑制活性)没有这种效果。莫那比拉韦是一种新的新冠病毒RNA聚合酶抑制剂,和以VeroE6细胞为基础的瞬时转染相比,它对VeroE6/Rep3细胞系有更好的抑制病毒活性潜力。他们的工作表明,该稳定转染新冠病毒复制子的细胞系是高通量筛选抗病毒药物的有力工具。
同年,Tony Wang等科学家[102]构建了用纳米荧光素酶取代S基因,新霉素磷酸转移酶基因取代E和M基因和携带NSP1蛋白K164A/H165A双突变的SARS-CoV-2的复制子(图2)。该复制子在电转进入到BHK-21细胞(携带稳定转染四环霉素诱导表达N蛋白的质粒)后,能瞬时表达纳米荧光素酶。在G418的选择压力下,得到了12个存活的细胞系。经20次传代后,纳米荧光素酶的表达没有丢失。同时细胞内存在SARS-CoV-2的RNA复制子的活跃复制。随后,他们利用该复制子筛选了273种不同化合物对病毒的抑制作用。这些化合物通过模拟筛选,能分别靶向新冠病毒的NSP5(3CLpro)、NSP3(PLpro)、NSP12(RdRP)、NSP15、NSP16和X区。在5 μmol/L的水平,通过纳米荧光素酶活性测定,9个化合物有50%的抑制效果。以瑞德西韦和GC376为阳性对照,分别检测了Darapladib、Genz-123346和JNJ-5207852等3个化合物的抑制效果。结果表明,3个化合物都表现出细胞类型特异的抑制活性。对6个细胞系进行GC376处理,发现它们的IC50值在5.9~13 μmol/L之间。总之,该论文作者巧妙地通过突变新冠病毒的NSP1蛋白降低新冠病毒对细胞的毒性,成功得到了稳定表达复制子并带有报告基因的细胞株,同时将它们应用到了大规模的抗新冠病毒药物库的筛选,开辟了一条筛选抗新冠病毒药物的新思路。
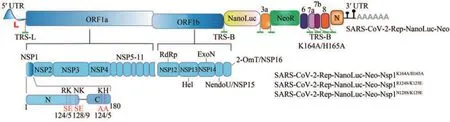
图2 新冠病毒复制子的优化[102]Fig. 2 Optimization of the SARS-CoV-2 replicon[102]
构建稳定转染复制子的细胞株,虽然花费的时间较长,但由于复制子能在细胞中持续的复制而不丢失,因而适合较长时间的复制、转录和翻译等的检测。同时,不同细胞批次之间结果比较稳定,一致性和重复性好,因而必将在高通量筛选抗病毒大化合物库的研究工作中得到越来越广泛的应用。
4 总结和展望
对于具有高度传染性的病毒来说,其相关的研究必须在具备生物安全三级或以上的专门实验室中进行,并且步骤烦琐,实验成本高,费时费力,这些不利因素阻碍了相关病毒学研究的发展,特别是在重大疫情突然来临时,不能及时和有效地应对疫情带来的危害。随着最近几十年现代病毒学研究技术的发展,科学家们通过删除一个或几个病毒关键结构基因,同时插入一个或几个报告基因和不同的表达调控元件,即构建病毒复制子,并用它来侵染相关细胞,用于病毒基因和宿主相关基因功能研究,以及抗病毒化合物的筛选。这样的好处是:一方面,病毒能在细胞内进行复制、转录和翻译;另一方面,由于缺乏一个或几个结构基因,不能持续产生完整的病毒粒子,从而导致了传染性的丧失。这些好处使得对病毒的相关基础研究和抗病毒药物研发可以在生物安全二级常规实验室中进行,具有安全、方便、实验成本低和快速的特点。这些病毒复制子理论和技术的发展为遏制重大疫情的传播和减少病毒的危害做出了重大贡献。
病毒复制子的构建技术在过去的几十年里得到了快速的发展。体外连接法、基于BAC质粒构建、酵母转化相关重组法和CPER法是目前构建新冠病毒复制子的四种主流方法。其中,体外连接法和基于BAC质粒构建法是最早应用的方法,其原理简单明了,但步骤多,过程冗长,往往需要多步的连接和转化过程,需要较高的实验水平和技巧。同时,部分病毒序列在大肠杆菌体内不稳定,增加了构建的难度。随后发展的酵母体内重组方法,所需的连接转化鉴定步骤少,仅一步就可以把多达十几个甚至几十个首尾具有相同序列的DNA片段和酵母-细菌穿梭载体连接起来,一次就可以得到全长的病毒复制子序列。这种方法的好处在于:第一,酵母对外源同源的DNA重组效率很高;第二,鉴定阳性酵母菌快速简单;第三,利用酵母-细菌穿梭人工染色体载体,通过电转的方法很容易将质粒从酵母转入大肠杆菌中,这极大地方便了大规模提取病毒复制子质粒,用于下一步的细胞转染实验。这些优点使得越来越多的研究人员采取该技术路线来构建新冠病毒和其他大病毒的复制子。最近发展的CPER方法只需要通过简单的PCR方法,就能将体外合成的若干具有首尾相同序列的DNA片段和载体环化,得到含有全长病毒DNA或cDNA的复制子,可直接用于细胞的转化。这种方法简单方便,不需要连接转化鉴定和细菌或酵母的培养以及质粒提取等冗长步骤,极大地节省了人力和时间。但由于PCR本身会带来一些突变,需要进行测序验证,并且对引物的选择要求比较高,增加了实验的难度。相信在未来这种方法也会得到更多的应用。
和瞬时转染相比,单周期感染复制子系统和稳定表达复制子细胞株具有更好的稳定性、一致性和重复性,因而能够为大规模高通量抗病毒药物筛选提供更好的平台。另外,单周期感染复制子系统使得研究新冠病毒侵染细胞膜上没有新冠病毒受体的细胞成为可能。
由于3针或以上疫苗接种的普及和充足的治疗药品,新冠病毒对中国人的身体健康危害已有所减轻。但是,由于新冠病毒的高传染性和高突变性并没有发生根本改变,在未来仍有可能流行,这将对一些免疫力低下的特殊人群健康造成严重影响。因此,相关的研究必须继续开展和深入探索。由于新冠病毒复制子具有精准地突变新冠病毒单个基因甚至单个核苷酸的能力,同时可以引入报告基因并在细胞中稳定表达,而且能在生物安全二级实验室操作等多个优点,因此在未来它仍将是科学家们用来研究新冠病毒的未知基因功能、致病机制以及筛选高特异和低副作用的抗新冠病毒药物的重要工具和手段。
总之,人工构建的新冠病毒复制子平台及其应用对研究新冠病毒的致病机制、基因功能、宿主因子的验证、病毒的变异和抗病毒药物的筛选提供了一种重要的手段,同时也对遏制新冠病毒的大规模传播、减少新冠病毒对人类健康的危害起到了不可或缺的作用。
