基于全长转录组的蚕豆WRKY基因家族分析及耐盐胁迫相关候选基因挖掘
2024-03-20周恩强姚梦楠王学军缪亚梅王永强汪凯华顾春燕魏利斌
周恩强, 周 瑶, 姚梦楠, 王学军, 赵 娜, 缪亚梅, 王永强, 薛 冬, 李 波,汪凯华, 顾春燕, 魏利斌
(江苏沿江地区农业科学研究所,江苏 南通 226012)
非生物胁迫,包括盐胁迫、干旱胁迫和低温胁迫等,限制了作物的生产力,并影响了植物中许多基因的表达[1],其中盐胁迫是制约全球农业发展和作物生产的最重要因素之一[2]。盐胁迫使植物遭受离子毒性、渗透胁迫和氧化胁迫,从而破坏各种细胞和生理过程。为了适应或抵抗盐胁迫,植物进化出几种策略,包括渗透调节(如渗透保护剂积累)、离子平衡 (Na+/K+平衡)和抗氧化(抗氧化酶的积累和活性的增强)[3]。植物对盐胁迫的适应性也可以通过复杂的信号通路改变许多基因的表达来实现,研究发现bZIP、WRKY、MYB等家族的转录因子通过结合特定的顺式作用元件形成一个复杂的调控网络,对植物的盐胁迫反应至关重要[4-6],这些转录因子过表达通常会增加植物对盐胁迫的适应性。
WRKY转录因子是植物中最大的转录因子家族之一,通过与保守的 DNA 结合位点W-box结合来调控基因的转录,在植物的生长发育、生物与非生物胁迫响应中发挥着多种作用[7]。所有的WRKY蛋白由1个或2个约60个氨基酸的WRKY保守结构域组成,其 N 端具有高度保守的WRKYGQK七肽序列,C 端具有 C2H2基序或 C2HC基序的锌指结构[8]。根据WRKY结构域的数量和类锌指基序的特征可将WRKY基因家族分为3组(I、II和III),第I组具有2个WRKY结构域,第II组具有1个WRKY结构域和C2H2基序,第III组具有1个WRKY结构域和C2HC基序[9]。目前WRKY基因家族已在多种植物中被鉴定出,其中拟南芥鉴定出72个WRKY基因[8],水稻中包含102个WRKY基因[10],大豆基因组中鉴定出197个WRKY基因[11],油茶中鉴定出89个WRKY基因[12],绿豆中鉴定出79个WRKY基因[13]。同时研究结果表明,WRKY转录因子基因在多种植物的盐胁迫响应中发挥着重要作用,例如,在拟南芥中过表达陆地棉GhWRKY34增强了拟南芥转基因植株的耐盐性[14];在烟草中过表达NbWRKY79增强了植株盐胁迫耐受性;在玉米中抑制ZmbZIP111的表达提高了玉米幼苗对盐胁迫的敏感性[15]。
WRKY转录因子基因参与植物生长发育的多个方面,在盐胁迫的多种不同响应途径中发挥重要作用,在模式植物拟南芥中,已发现多个受盐胁迫调控的WRKY基因。Babitha等[16]研究发现共表达AtbHLH17和AtWRKY28能够增强拟南芥对氯化钠、甘露醇胁迫和氧化胁迫的耐受性。在盐胁迫和脱落酸(ABA)处理下,相比于野生型,Atwrky66突变体表现出对ABA和盐胁迫更高的敏感性[17]。Atwrky25突变体与Atwrky33突变体的盐胁迫敏感性与野生型没有太大差异,但过表达AtWRKY25或AtWRKY33增加了拟南芥对氯化钠胁迫的耐受性[18]。研究发现JMJ15通过调控AtWRKY46和AtWRKY70的表达水平来增强其耐盐性,并证明AtWRKY46和AtWRKY70在盐胁迫中起负调控作用[19]。在盐胁迫下,AtWRKY1的表达被诱导,且AtWRKY1功能缺失导致了突变体拟南芥的盐敏感性增加[20]。Li等[21]利用基因编辑技术(CRISPR/Cas9)对AtWRKY3和AtWRKY4进行了编辑,发现突变体对盐和茉莉酸甲酯(MeJA)胁迫的耐受性降低。与野生型相比,过表达AtWRKY30的拟南芥植株对氧化胁迫和盐胁迫表现出更强的耐受性[22]。AtWRKY9通过调控AtCYP94B3和AtCYP86B1来控制软木脂的沉积,从而增加拟南芥的耐盐性[23]。Chen等[24]通过对AtWRKY18、AtWRKY40和AtWRKY60进行单突、双突、三突和过表达分析发现,AtWRKY18和AtWRKY60对抑制种子萌发和根系生长有积极作用,同时2个WRKY基因也提高了拟南芥对盐胁迫和渗透胁迫的敏感性。拟南芥AtWRKY8主要在根中表达,并在盐处理后显著上调表达,AtWRKY8的功能缺失使拟南芥在盐胁迫环境下受到抑制作用,表现出萌发延迟,并且抑制萌发后的生长发育[25]。在盐胁迫条件下,与野生型相比,Atwrky11和Atwrky17突变体的萌发速度较慢,根系生长受损,表现出盐胁迫敏感性[26]。
蚕豆(ViciafabaL.)属于豆科野豌豆属一年生或越年生草本植物[27],因其较高的营养价值和有效的生物固氮作用在作物生产中占据着重要地位。随着人们生活质量的不断提高,市场对蚕豆的需求呈增加和多样化趋势[28],由于中国耕地资源有限,粮、菜、油争地矛盾突出,现有的蚕豆种植面积已无法满足蚕豆生产发展的需求,可以通过挖掘基因资源、培育适宜盐碱地种植的蚕豆新品种,从而扩大蚕豆种植面积来满足市场需求。
1 材料与方法
1.1 试验材料
本研究使用的蚕豆品种启豆2号种植于江苏沿江地区农业科学研究所种植基地。采集根(盛花期)、茎叶(盛花期)、花(盛花期)、种子(开花后20 d、30 d、40 d)、果皮(开花后20 d、30 d、40 d)等样本保存于液氮中备用。委托北京百迈客生物科技有限公司进行二代转录组(9个样本,3次重复,27个样品)和全长转录组(9个样本RNA等量混合)测序、检测、分析等。
1.2 全长转录组数据处理
根据条件fullpasses(全票)≥3且序列准确性大于0.9从原始序列提取CCS(Circular consensus sequencing read)序列,并对CCS序列进行校正。检测CCS序列中是否包含5′引物、3′引物及poly A尾,3个都包含的为全长非嵌合序列(Full-length no chimera, FLNC)。使用SMRTLink软件中的IsoSeq模块将全长非嵌合序列中相似的序列聚类到一簇(Cluster),每个Cluster得到一个一致序列(Consensus isoform),通过minimap2将得到的校正后的一致序列与蚕豆Tiffany参考基因组(https://projects.au.dk/fabagenome/genomics-data)进行序列比对(设置参数-ax splice -uf --secondary=no -C5),使用cDNA Cupcake软件对比对结果去冗余,过滤Identity(一致性)小于0.9、Coverage(优势度)小于0.85的序列,合并仅5′端外显子有差异的比对,最终得到非冗余转录本。
1.3 VfWRKY基因家族成员的鉴定
本研究以蚕豆全长转录组为研究对象。在 pfam 蛋白质家族数据库(http//pfam. xfam. org/)中查找获取WRKY基因家族保守结构域的序列号(PF03106),并下载其对应的隐马尔可夫模型文件[29],利用TBtools软件在蚕豆全长转录组蛋白质序列中初步检索具有WRKY保守结构域的WRKY转录因子。分别从拟南芥(https://www.arabidopsis.org/index.jsp)和PlantTFDB v5.0(http://planttfdb.gao-lab.org/index.php)网站下载拟南芥和蒺藜苜蓿的WRKY家族基因序列,并将其比对到蚕豆全长转录组中获取序列相似性最高的基因。将获取的WRKY蛋白质序列提交到NCBI CDD(https://www. ncbi. nlm. nih. gov/cdd)数据库,明确是否含WRKY保守结构域,并剔除重复、冗余和注释不完整的序列,保留下来的转录因子即为蚕豆WRKY家族成员。
1.4 VfWRKY基因家族成员理化性质及亚细胞定位分析
利用TBtools软件计算蚕豆WRKY家族成员编码蛋白质的氨基酸大小、相对分子质量、等电点、不稳定指数、脂肪系数和亲疏水性。利用在线网站Cell-PLoc 2.0(http://www.csbio.sjtu.edu.cn/bioinf/Cell-PLoc-2/)对WRKY家族成员进行亚细胞定位预测。
1.5 VfWRKY基因家族成员保守结构域及系统进化分析
提交拟南芥和蚕豆WRKY家族基因蛋白质序列到MEGA 7. 0软件中进行多重比对,将比对结果使用最大似然法(Maximum likelihood,ML)构建进化树。提交进化树参数到网站Evolview(https://evolgenius.info//evolview-v2/#login)上美化进化树[30],并根据拟南芥WRKY基因家族分类方法进行分组[8]。单独提交蚕豆WRKY家族基因蛋白质序列到MEGA 7. 0软件中进行多重比对,使用最大似然法构建进化树,将进化树参数和WRKY家族基因保守结构域信息输入到TBtools软件中进行数据可视化分析[31]。
1.6 VfWRKY基因家族成员染色体分布及Motif鉴定
将113个WRKY基因家族成员的基因编号提交到TBtools软件中,即可将每个基因定位到相应的染色体上。使用在线软件MEME(https://meme-suite. org/meme/tools/meme)预测蚕豆WRKY基因的保守基序[32],Motif最大值设置为10。下载Motif分析文件与蚕豆进化树参数共同提交到TBtools软件中进行数据可视化分析。
1.7 VfWRKY基因家族成员GO富集和KEGG通路分析
利用BLAST比对工具,将蚕豆WRKY家族转录本与基因本体(Gene Ontology,GO)(http://www.geneontology.org/)和KEGG(https://www.kegg.jp/)公共数据库进行比对,从而得到WRKY蛋白功能注释信息,并将该注释信息提交到百迈客云在线分析平台(https://international.biocloud.net/zh/software/tools/)进行可视化分析。
除了现有研究已经涉及的高唐神女、观音、阿尼玛、鱼、力、死亡与再生、大地母亲与智慧老人等原型,沈从文的小说中还存在许多值得探究的原型。比如,《媚金·豹子与那羊》《月下小景》等小说是沈从文根据湘西神话故事改编而来的,其中涉及“难题求婚”的文学母题。又如,沈从文笔下有许多雄健俊美的男子,《渔》中的孪生兄弟、《边城》中的天保、傩送兄弟等,这类反复出现的男子形象,也可能蕴藏着某种原型。所有这些都有待学者们的深入探讨。
1.8 蚕豆盐胁迫候选WRKY基因的挖掘
通过文献查找模式植物拟南芥中与盐胁迫相关的WRKY基因,下载其蛋白质序列与蚕豆WRKY蛋白序列进行同源比对,从而获得蚕豆盐胁迫相关候选WRKY基因。利用全长转录组表达量数据对盐胁迫相关候选WRKY基因进行表达模式分析。
2 结果与分析
2.1 全长转录组数据统计
通过 PacBio高通量测序技术对启豆2号的根、茎叶、花、种子、果皮等组织进行全长转录组测序,共获得53.84 Gb原始数据。全长转录组测序获得474 220条环形一致性序列 (CCS),平均长度1 936 bp,分布于1 000~1 499 bp的CCS数量最多,为121 790条(图1a);全长非嵌合序列(FLNC)有390 912条,平均长度1 646 bp,分布于500~999 bp的FLNC数量最多,为100 818条(图1b)。对全长非嵌合序列进行聚类和去冗余分析,最终得到58 885条转录本序列,其中新转录本数量为42 019个,平均长度为2 209 bp,分布于81~9 800 bp,主要集中在500~2 499 bp区间内,1 000~1 499 bp数量最多,为6 696个转录本(图1c);我们对新发现的42 019个转录本进行序列结构分析,预测出26 438条完整开放阅读框(Open reading frame,ORF)序列,平均长度为1 038 bp,分布于500 bp以下的ORF数量最多,为9 257条(图1d)。
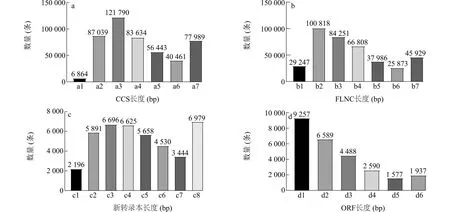
a:环形一致性序列(CCS)长度分布;b:全长非嵌合序列(FLNC)长度分布;c:新转录本长度分布;d:开放阅读框(ORF)长度分布。a1:<500;a2:500~999;a3:1 000~1 499;a4:1 500~1 999;a5:2 000~2 499;a6:2 500~2 999;a7:≥3 000;b1:<500;b2:500~999;b3:1 000~1 499;b4:1 500~1 999;b5:2 000~2 499;b6:2 500~2 999;b7:≥3 000;c1:<500;c2:500~999;c3:1 000~1 499;c4:1 500~1 999;c5:2 000~2 499;c6:2 500~2 999;c7:3 000~3 499;c8:≥3 500;d1:<500;d2:500~999;d3:1 000~1 499;d4:1 500~1 999;d5:2 000~2 499;d6:≥2 500。
2.2 VfWRKY转录因子家族的鉴定及理化性质分析
基于蚕豆全长转录组数据,在TBtools软件中使用pfam程序搜索出121个WRKY转录本,经数据库比对以及去除冗余和缺失序列的转录因子,最终鉴定筛选出113个WRKY转录本,并重新命名为VfWRKY1~VfWRKY113,其中相对蚕豆Tiffany参考基因组新发现WRKY转录本31个(以PB命名)。VfWRKY转录因子基因家族编码的蛋白质的理化性质见表1,蚕豆113个WRKY转录因子编码的氨基酸数目最小为VfWRKY61(153 aa),最大为VfWRKY36(737 aa);相对分子质量为17 729.49~81 727.72;等电点为4.84~9.87,76个蛋白质等电点小于7.00,为酸性蛋白质,37个蛋白质等电点大于7.00,为碱性蛋白质;不稳定指数为29.87~67.85,7个蛋白质不稳定指数小于40.00,为稳定蛋白质,106个蛋白质不稳定指数大于40.00,为不稳定蛋白质;脂肪系数为45.60~81.99;平均亲疏水性为-1.42~-0.40,均小于 0,表明蚕豆113个WRKY家族蛋白质均属于疏水性蛋白质。亚细胞定位预测发现113个WRKY家族蛋白质均定位于细胞核中,间接证明113个WRKY基因作为转录因子调控下游基因的表达。
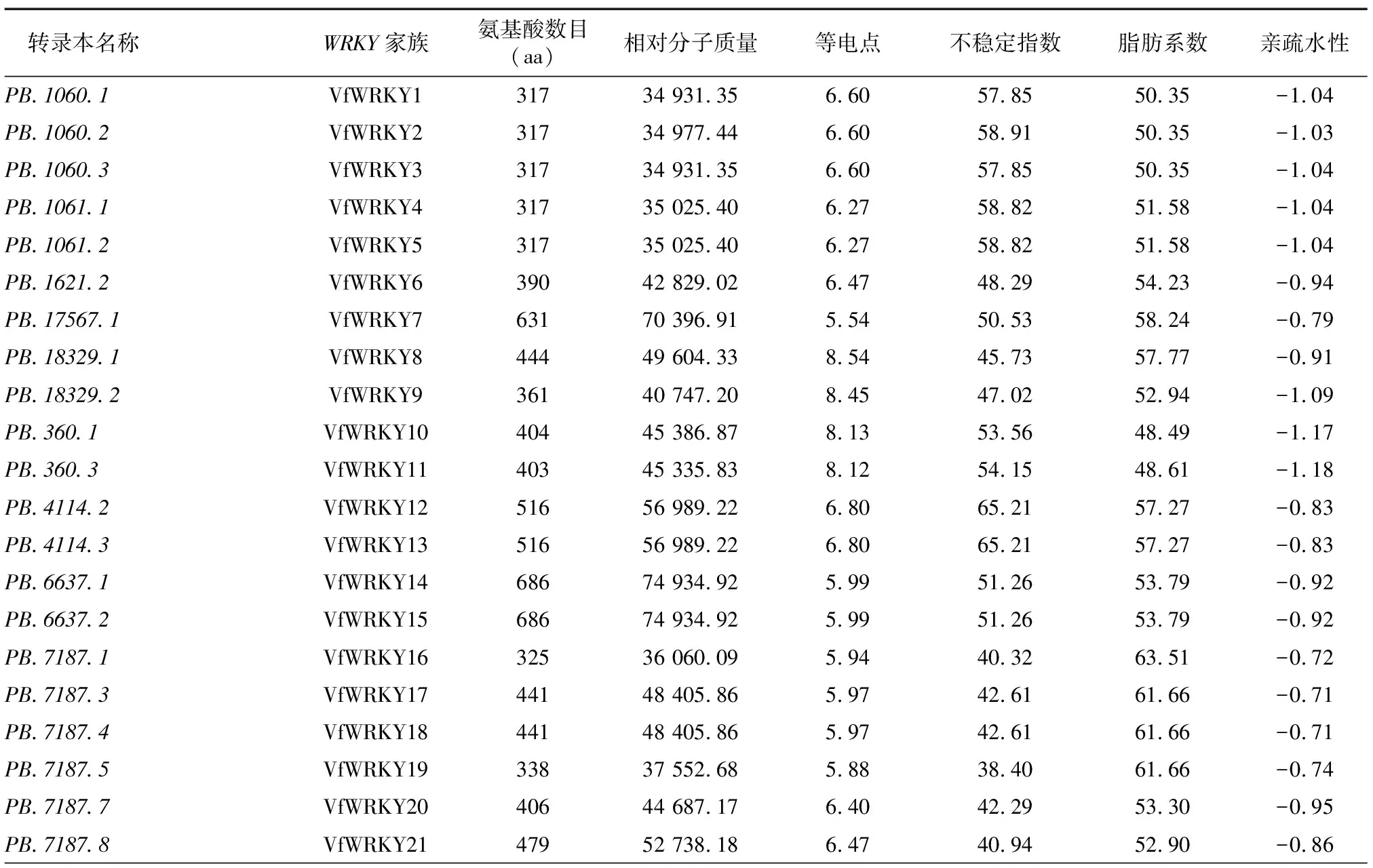
表1 蚕豆WRKY 转录因子家族成员信息
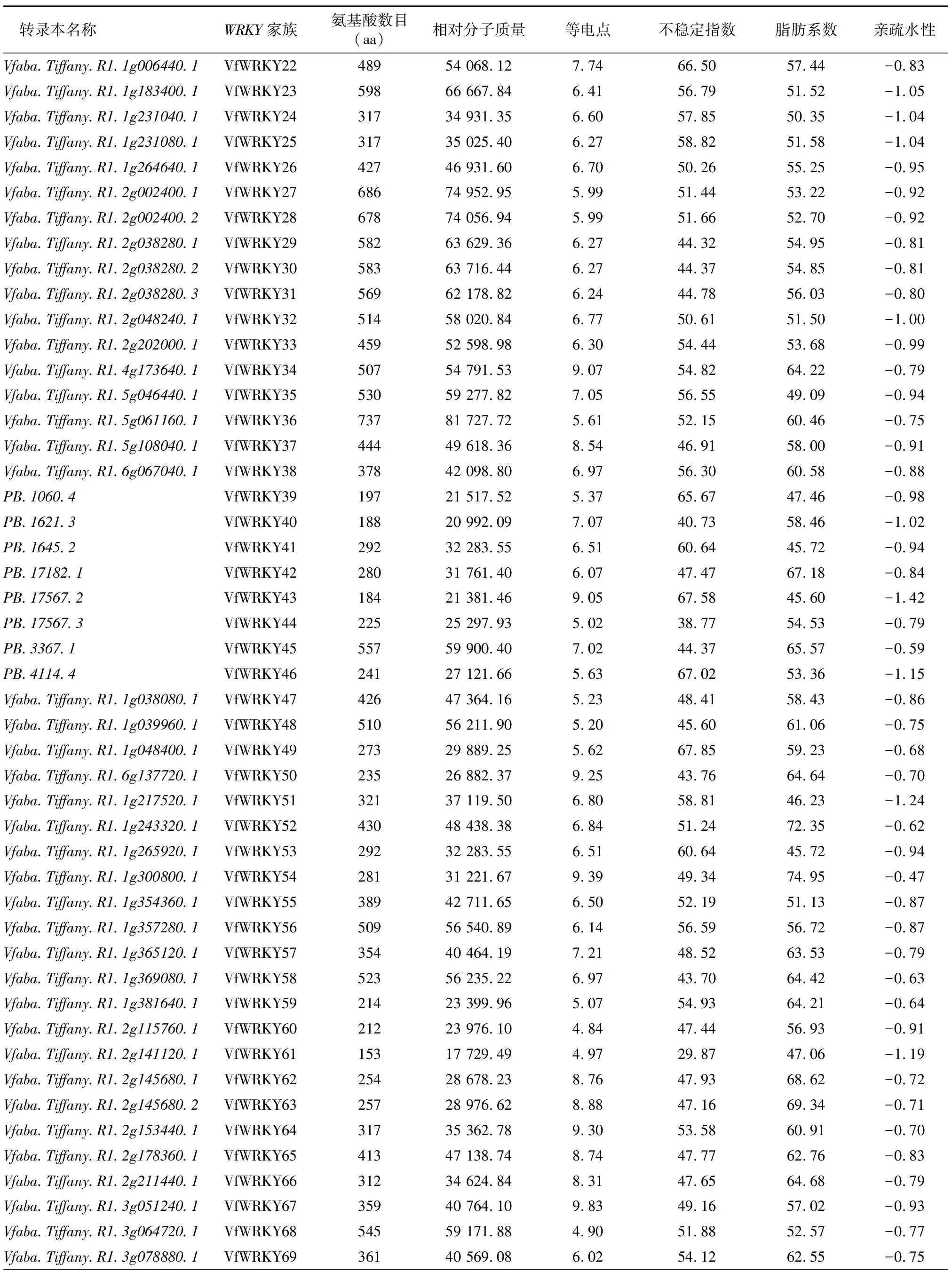
续表1 Continued 1

续表1 Continued 1
2.3 VfWRKY家族成员染色体分布及系统进化分析
为更好地了解蚕豆WRKY基因家族系统发育关系,我们利用最大似然法构建了71个拟南芥WRKY蛋白和113个蚕豆WRKY蛋白的系统发育树,并根据Eulgem等[8]的分类方法将蚕豆WRKY家族成员分为group 1~group 3,group 1具有2个WRKY结构域,group 2具有1个WRKY结构域和C2H2基序,group 3具有1个WRKY结构域和C2HC基序。系统发育树分析结果显示,group 1、group 2、group 3分别包含38个(VfWRKY1~VfWRKY38)、61个(VfWRKY39~VfWRKY99)和14个(VfWRKY100~VfWRKY113)VfWRKY基因,group 3包含的基因数目最少,且与拟南芥13个WRKY基因聚在同一亚家族中。同时我们发现除group 1中VfWRKY16~VfWRKY19与group 2中VfWRKY基因聚在同一分支上外,其他group 1和group 2WRKY家族成员与拟南芥该亚家族成员都能够很好地聚在同一分支上(图2)。

图2 蚕豆和拟南芥WRKY基因系统发育分析
我们将VfWRKY16~VfWRKY19与VfWRKY29(5个基因同为Vfaba.Tiffany.R1.2g038280的转录本)进行多序列比对发现5个基因的第一个WRKY结构域都为WRKYGQK-C2-HDH(完整),但VfWRKY16、VfWRKY17、VfWRKY18和VfWRKY19的第二个WRKY结构域只有WRKYGQK-(VfWRKY29的第二个WRKY结构域具有完整的WRKYGQK-C2-H2结构),所以这4个基因聚集在group 2分支是由于第二个结构域缺少C2-H2序列(图3)。

标记处表示WRKY结构域氨基酸序列。
由于所使用的软件或数据本身具有局限性,导致所选参考基因组注释往往不够精确,这样就有必要对原有注释的基因结构进行优化。我们通过gffcompare软件与蚕豆Tiffany参考基因组注释进行比较,将已知基因和转录本保留原身份标识号(ID),将新基因和新转录本保留PB格式的ID添加到注释中,最终我们得到包含新基因和新转录本的优化参考基因组注释文件。利用优化参考基因组注释文件提取位置信息绘制WRKY家族成员在染色体上的物理分布(图4)。蚕豆由12条染色体组成,每两条染色体为1对,共6对,其中染色体1包含染色体1L和染色体1S。113个WRKY转录本不均匀地分布在7条染色体上,染色体1拥有的WRKY家族成员最多,为38个,其中染色体1L有29个,染色体1S有9个; 染色体2拥有WRKY家族成员 24个,染色体5拥有WRKY家族成员19个,染色体4拥有WRKY家族成员13个,染色体6 拥有WRKY家族成员11个,染色体3拥有WRKY家族成员数量最少,为8个。
2.4 VfWRKY家族保守结构域和保守基序鉴定
113个VfWRKY蛋白包含3种保守结构域,分别为WRKY、Plant_zn_clust、PTZ00265。其中VfWRKY54、VfWRKY64、VfWRKY67、VfWRKY72、VfWRKY77、VfWRKY78和VfWRKY82含有WRKY和Plant_zn_clust结构域,并且7个基因聚在同一分支上;VfWRKY14、VfWRKY15、VfWRKY27和VfWRKY28含有WRKY和PTZ00265结构域,其余102WRKY基因都只包含WRKY结构域(图5)。同时我们发现group 2和group 3中的61个和14个WRKY转录本都只含有1个WRKY保守结构域,并且分别聚集在同一分支上;group 1的38个WRKY转录本都含有2个WRKY保守结构域,但VfWRKY16、VfWRKY17、VfWRKY18和VfWRKY19的第二个WRKY结构域长度不完整(图5)。
在一个基因家族中,基因的保守基序可以反映该基因家族成员之间的进化关系和功能。我们利用在线网站MEME分析了VfWRKY基因家族的保守基序,并使用TBtools进行了可视化分析(图6)。VfWRKY基因家族共含有10个Motif,其中不同的WRKY基因含有Motif的数量为2~10个,VfWRKY39和VfWRKY44只含有2个Motif,VfWRKY12、VfWRKY13、VfWRKY22、VfWRKY29、VfWRKY30和VfWRKY31含有的Motif最多,为10个。通过序列查询发现Motif 1和Motif 3与WRKY结构域相对应,group 2和group 3中有71个WRKY转录本含有Motif 1,只有VfWRKY39、VfWRKY43、VfWRKY44和VfWRKY46含有Motif 3;group 1中37个基因同时含有Motif 1和Motif 3,只有VfWRKY34含有2个Motif 1。每个WRKY亚家族都具有相对稳定的Motif,其中group 1亚家族有33个基因(在group 1中占比86.8%)同时含有Motif 3、Motif 6、Motif 7、Motif 1、Motif 4、Motif 2和Motif 5;group 2亚家族被分为2个分支,分支1有29个基因(在group 2中占比47.5%)同时含有Motif 1、Motif 4、Motif 2和Motif 5,分支2有27个基因(在group 2中占比44.3%)同时含有Motif 7、Motif 1、Motif 4、Motif 2和Motif 5;group 3亚家族所有基因都含有Motif 1、Motif 4和Motif 2。VfWRKY16、VfWRKY17、VfWRKY18和VfWRKY19的Motif数量与保守结构域长度相对应,相对group 1亚家族成员缺少了Motif 4、Motif 2和Motif 5(图6)。

图4 蚕豆WRKY基因在染色体上的分布

图5 蚕豆WRKY家族成员保守结构域分析

图中色块上1~10表示motif序号。
2.5 VfWRKY家族成员GO功能注释及KEGG通路分析
将VfWRKY家族的113个WRKY转录本与GO 数据库进行比对分析,结果显示,113个WRKY转录本都得到了注释(图7a)。按照GO功能分类方式,将113个WRKY转录本分为生物过程(Biological process)、细胞组分(Cellular component)和分子功能(Molecular function)3大类。3个大类可细分为20个二级分类,其中生物过程包含的二级分类最多,为16个不同的亚类,主要包括转录调控(DNA和RNA模板转录)、代谢过程的调控(大分子、细胞、化合物和RNA等)、生物合成过程的调控(细胞、大分子和RNA等);分子功能注释基因占比最多,为112个WRKY转录本,主要包括DNA结合转录因子活性和序列特异性DNA结合2个功能;细胞组分包含的二级分类最少,主要为细胞核(29个转录本)。
在KEGG 数据库对VfWRKY家族的113个WRKY转录本进行通路富集分析,结果显示有98个VfWRKY转录本得到KEGG通路富集(图7b)。VfWRKY基因主要富集在植物MAPK信号通路、植物与病原菌相互作用和剪接体3个通路中,其中植物与病原菌相互作用通路WRKY基因最多,为84个(占比85.71%),其次是植物MAPK信号通路,为40个(占比40.82%),剪接体通路最少,为13个(占比13.27%)。

P值、Q值表示差异显著性检验指标。图a中,柱长度对应P值的对数,柱后的数字表示基因数。
为了探究蚕豆WRKY家族成员表达模式,我们利用WRKY基因在根、茎叶、花、种子(开花后20 d、30 d、40 d)、果皮(开花后20 d、30 d、40 d)中的表达信息,构建了WRKY基因表达图谱(图8)。分析表达图谱可知,WRKY基因家族在9个样本中的表达模式可分为5组(Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅴ):Ⅰ组主要在果皮中高表达;Ⅱ组主要在种子中高表达;Ⅲ组在9个样本中低表达或不表达;Ⅳ组主要在根中表达且在其他组织中低表达或不表达,并且在根中高表达的WRKY基因占比较高(46.0%);Ⅴ组在多个组织中均有表达。

R:根;L:茎叶;F:花;S20、S30、S40表示开花后20 d、30 d、40 d的种子;P20、P30、P40表示开花后20 d、30 d、40 d的果皮。
2.6 蚕豆盐胁迫候选WRKY基因的挖掘
通过查阅已有的拟南芥WRKY基因家族报道,我们发现AtWRKY1、AtWRKY3、AtWRKY4、AtWRKY8、AtWRKY9等16个基因与盐胁迫相关,其中12个基因为耐盐基因,AtWRKY18、AtWRKY46、AtWRKY60、AtWRKY70为盐敏感基因(表2)。将拟南芥中与盐胁迫相关的16个WRKY基因比对到蚕豆WRKY基因家族中,在蚕豆中发现了14个候选同源基因,分别为Vfaba.Tiffany.R1.1g183400、Vfaba.Tiffany.R1.2g038280、Vfaba.Tiffany.R1.3g164760、Vfaba.Tiffany.R1.2g048240、Vfaba.Tiffany.R1.4g037840、Vfaba.Tiffany.R1.6g074800、Vfaba.Tiffany.R1.3g078920、Vfaba.Tiffany.R1.1g060200、Vfaba.Tiffany.R1.2g211440、Vfaba.Tiffany.R1.5g003240、Vfaba.Tiffany.R1.1g006440、Vfaba.Tiffany.R1.2g102760、Vfaba.Tiffany.R1.6g076880和Vfaba.Tiffany.R1.5g049280,其中AtWRKY3和AtWRKY4共同比对到Vfaba.Tiffany.R1.1g006440,AtWRKY11和AtWRKY17共同比对到Vfaba.Tiffany.R1.3g164760,且AtWRKY3、AtWRKY4、AtWRKY11和AtWRKY17都为耐盐基因。
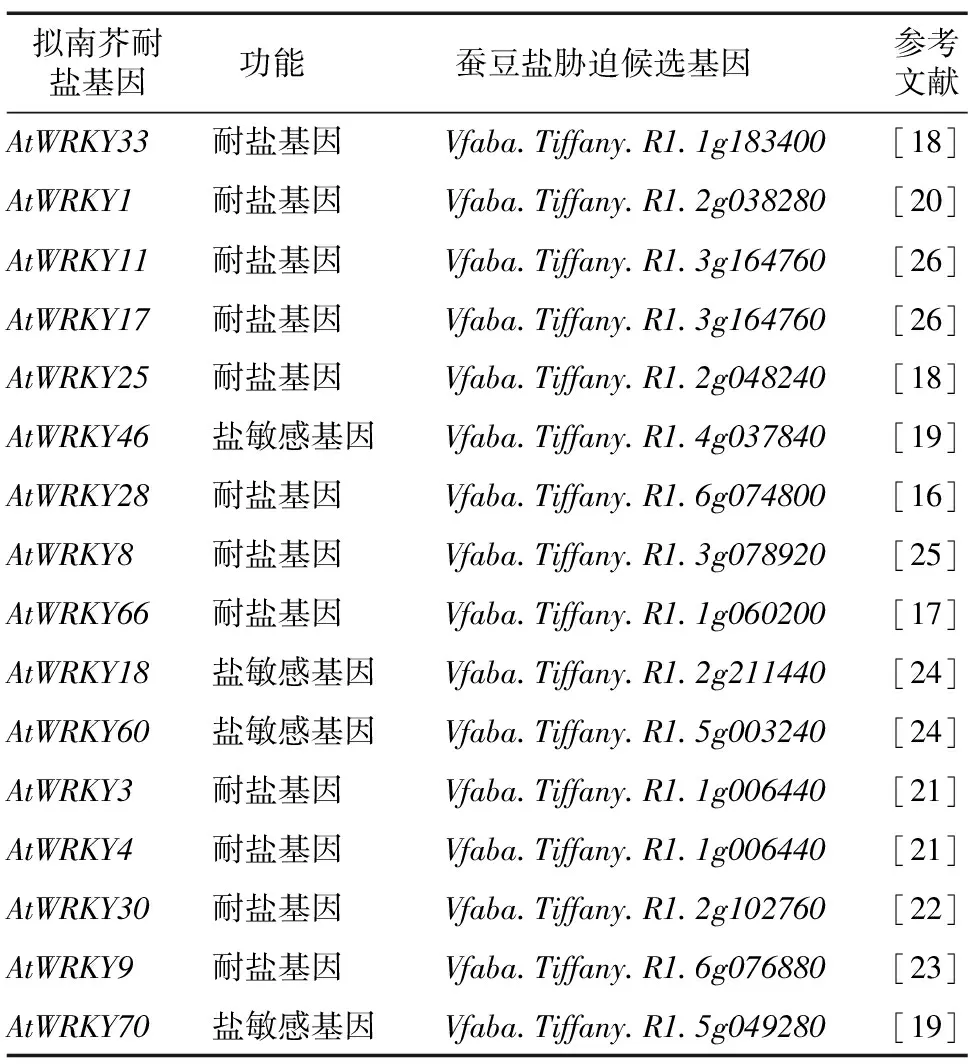
表2 蚕豆盐胁迫候选WRKY基因
我们对启豆2号的根、茎叶、花、种子(开花后20 d、30 d、40 d)、果皮(开花后20 d、30 d、40 d)的27个样品进行了转录组分析,利用转录组数据对蚕豆14个盐胁迫相关候选WRKY基因进行了表达模式分析(图9)。Vfaba.Tiffany.R1.1g183400、Vfaba.Tiffany.R1.3g164760、Vfaba.Tiffany.R1.2g048240、Vfaba.Tiffany.R1.4g037840、Vfaba.Tiffany.R1.6g074800、Vfaba.Tiffany.R1.2g211440、Vfaba.Tiffany.R1.5g003240、Vfaba.Tiffany.R1.2g102760、Vfaba.Tiffany.R1.6g076880主要在根中高表达,在其他组织的表达量较低或不表达。Vfaba.Tiffany.R1.1g006440在多个组织中均有表达,并在开花后40 d果皮中表达量最高;Vfaba.Tiffany.R1.1g060200在多个组织中表达量较低或不表达;Vfaba.Tiffany.R1.5g049280在开花后20 d果皮中表达量最高。Vfaba.Tiffany.R1.1g183400、Vfaba.Tiffany.R1.2g038280、Vfaba.Tiffany.R1.2g048240、Vfaba.Tiffany.R1.2g102760、Vfaba.Tiffany.R1.2g211440、Vfaba.Tiffany.R1.3g078920、Vfaba.Tiffany.R1.3g164760、Vfaba.Tiffany.R1.4g037840和Vfaba.Tiffany.R1.5g003240在果皮中的表达量均先升高后下降(图9)。

R:根;L:茎叶;F:花;S20、S30、S40表示开花后20 d、30 d、40 d的种子;P20、P30、P40表示开花后20 d、30 d、40 d的果皮。
3 讨 论
蚕豆是具有较高营养价值和生物固氮作用的食用豆类,由于缺乏参考基因组序列,使得蚕豆的基础研究进展缓慢。2023年Jayakodi等[33]通过高通量测序,组装了第一个蚕豆高质量、染色体规模的基因组,并公布了37 065个基因的序列信息。全长转录组测序(三代高通量测序技术)能够直接获得高质量全长转录本,高质量全长转录本既可以为无参基因组的二代测序数据拼接提供参考,也可以完善基因组的基因数量。本研究首次对蚕豆的根、茎叶、花、种子(开花后20 d、30 d、40 d)、果皮(开花后20 d、30 d、40 d)等9个样本进行了三代转录组测序,获得58 885 条转录本序列,与蚕豆基因组相比,发现新转录本42 019个。本研究构建的全长转录组为后续进一步开展蚕豆遗传研究提供了丰富的数据基础。
WRKY转录因子基因是植物特有的转录因子基因,已有研究结果表明,WRKY转录因子基因能够调控植物生长发育,例如,调控种子休眠、萌发[34],调控幼苗的形态发生[35],以及调节植物的开花时间等[36]。除了这些功能外,WRKY基因在响应非生物胁迫(干旱、高温、高盐等)[37]和生物胁迫(病原菌[38]和昆虫[39]等)方面也具有非常重要的作用。本研究首次利用全长转录组测序对蚕豆WRKY家族进行研究,并鉴定出113个VfWRKY基因。不同物种间系统进化树的建立,有助于更加准确地研究物种中未知家族成员的功能,我们根据进化关系,将蚕豆113个WRKY基因分成3组,其中group 2成员最多,为61个。同时我们对VfWRKY家族进行保守结构域和保守基序分析发现,group 1中的4个基因(VfWRKY16、VfWRKY17、VfWRKY18、VfWRKY19)具有2个WRKY保守结构域,但却与group 2亚家族聚集在同一分支,通过序列比对,发现是由于第二个WRKY结构域缺少C2-H2序列造成的,需要通过优化蚕豆参考基因组后来完善其序列。
VfWRKY基因家族主要富集在植物MAPK信号通路、植物与病原菌相互作用和剪接体3个通路中。刘晨等[40]通过分析前人的研究发现MAPK信号通路能够响应干旱胁迫、盐胁迫、极端温度及营养匮乏等非生物胁迫,并在植物抗逆过程中扮演重要角色,这与本研究的结果相符。同时WRKY基因在植物与病原菌相互作用通路中也具有多种功能,研究发现AtWRKY25和AtWRKY33既能调控植物与病原菌的相互作用[41-42],也能增强植物抵抗盐胁迫的能力[24]。根据拟南芥WRKY基因家族已知基因功能,通过同源比对,在VfWRKY基因家族中发现14个候选基因可能与盐胁迫相关,其中部分基因同时富集在植物与病原菌相互作用和植物MAPK信号通路中。本研究的下一步工作将对14个候选基因进行功能验证,探索其与盐胁迫的相关性。
