五氟吡啶激发态非绝热弛豫过程中的分子结构*
2024-03-19李多多张嵩
李多多 张嵩
1) (中国科学院精密测量科学技术创新研究院,波谱与原子分子物理国家重点实验室,武汉 430071)
2) (中国科学院大学,北京 100049)
利用量子化学计算研究了五氟吡啶分子的激发态非绝热弛豫路径中一些关键点的分子结构和能量.计算确定了五氟吡啶分子基态及两个最低激发态的结构和相应电子态的垂直和绝热激发能,其中基态是具有C2v 对称性的平面结构,而激发态结构为平面外畸变的半船型结构.同时确定了3 个锥形交叉S2/S1,S1/S0,S2/S0的拓扑结构和能量.在分支空间中,锥形交叉S2/S1,S1/S0,S2/S0 的结构都是尖峰不对称结构,分别为船型、半船型和椅式结构,其能量分别为6.39,5.16 和8.51 eV.计算结果表明五氟吡啶分子的非辐射弛豫主要是S2 态上的波包经锥形交叉S2/S1 快速内转换到S1态,再通过S1/S0 弛豫到基态的路径,而直接通过S2/S0 衰减到基态的概率较小.
1 引言
Born-Oppenheimer (BO)近似假设了分子中原子核和电子的运动可以分离,波函数可以分解为电子和核的两部分[1].然而,由于电子和核之间存在较强的耦合,BO 近似会失效,电子态之间发生非绝热跃迁.非绝热跃迁会导致波包从在不同激发态之间发生快速转换.激发态非绝热耦合形成的锥形交叉在非绝热跃迁过程中表现出独特的动态特性[2–6],为分子提供有效的快速弛豫途径,在分子激发态的非绝热动力学过程中具有非常重要的作用[7–11].
氟是电负性较强的元素,由于其强吸电子能力可降低材料的氧化速率,这使得含有氟原子取代的有机材料分子在航空航天工业和生物医学中通常具有很好性能和应用前景[12–17].五氟吡啶分子是重要的全氟芳香烃化合物之一,氟原子及杂原子的引入会对分子的结构和电子分布产生明显的影响,使得五氟吡啶分子具有特殊的物理化学性质,对分子受激后的反应机理及弛豫过程产生影响.由于原子或自由基引入芳香族结构会增加原子核的权重并导致激发能量的降低,氟代苯的吸收谱相对于苯的吸收谱出现明显的红移[18].此外,氟原子引入芳香环或杂环中会增强π 键及其反键特征.且随着氟原子数目的增加,C—F 键的键长逐渐变短,使得氟取代的芳香族化合物的分子结构会更稳定、芳香性更强[19].这些结构变化引起的分子物理化学性质的差异与氟原子的强电负性密切相关,氟取代导致了电子密度从芳香环向氟原子扩展[20–22].
氟苯和五氟吡啶分子的飞秒时间分辨的光电子影像研究,揭示了ππ*态与相邻的πσ*态的振动相互作用[20,21].研究者观察到11B2(ππ*)态和11A2(πσ*)态之间有9 ps 的振动相干性,分子在光激发后发生的弛豫动力学过程受到了振动耦合的影响.这些振动耦合驱动了五氟吡啶分子电子态之间的非绝热耦合,并通过锥形交叉快速弛豫,但是关于五氟吡啶分子的锥形交叉结构和能量等相关信息并不全面,尤其是不同电子态之间由非绝热耦合形成的锥形交叉结构对弛豫过程的影响.本工作基于密度泛函方法和多参考自洽场方法研究了五氟吡啶分子基态和两个低电子激发态的结构和能量,以及分子被激发到不同电子态后在弛豫演化路径中的锥形交叉的结构特性,通过分析弛豫演化路径中所有关键点的结构和能量,讨论了五氟吡啶分子激发态的经锥形交叉后的无辐射弛豫过程的机制.
2 计算方法
本文使 用B3LYP,M062X,SA-CASSCF 等方法结合6-311G*基组,对五氟吡啶分子的S0态、S1态和S2态的结构进行优化.其中,SA-CASSCF方法选取了由3 个π 轨道、3 个π*轨道、1 个σ*轨道以及孤立氮原子的一个n轨道组成的8 个轨道和8 个电子的活化空间,标记为SA-CASSCF(8,8).此外,采用M062X 方法结合6-311G*基组对S2态进行几何优化时,结构不收敛,而采用6-31G*基组对S2态进行优化以获得稳定结构.采用B3LYP,M062X,SA-CASSCF(8,8)和CASPT2 方法计算了五氟吡啶分子S1态和S2态的垂直激发能(vertical excitation energies VEEs)以及绝热激发能(adiabatic excitation energies,AEEs),其中S2态的AEE 是在M062X/6-311G*水平下进行.在SACASSCF(8,8)/6-31G*水平对锥形交叉S2/S1的几何结构进行优化,同时,采用SA-CASSCF(8,8)/6-311G*水平对锥形交叉S1/S0和S2/S0的结构进行了优化,并获得3 个锥形交叉点的相应能量.在B3LYP/6-311G*水平下,利用NEB (nudged elastic band)方法计算了势能面上连接S2态和锥形交叉S2/S1之间的最小能量路径.本工作为证实所有优化得到的结构为全局最优结构,在每个优化后的构型下还进行了频率计算,确保没有虚频.此外,对AEEs 都进行了零点能量校正.
本工作中,密度泛函和多参考自洽场方法分别使用Gaussian09[23]和Molpro[24]计算软件进行.NEB 最小能量路径计算使用Ocar 软件进行[25].不同激发态的优化结构和锥形交叉点的结构由Gaussview[26],Multiwfn[27]和Molden[28]等可视化软件生成.
3 结果与讨论
3.1 五氟吡啶分子的几何结构
利用B3LYP,M062X 和SA-CASSCF(8,8)等方法结合6-311G*基组对基态S0态的几何结构进行了优化,如图1 所示.计算结果表明,在不同的方法下优化获得的基态的几何结构相似,都是具有C2v对称性的平面结构.在不同计算水平下,五氟吡啶分子S0态的C—C 键和C—F 键的键长分别在1.38—1.39 Å和1.31—1.33 Å范围.而五氟吡啶分子S0态的C—N 键的键长都是1.31 Å,略短于C—C 键的键长.

图1 (a)五氟吡啶分子的结构和对应的原子序数;(b)—(d) 利用B3LYP,M062X 和SA-CASSCF(8,8)方法下获得的S0 态的几何结构Fig.1.(a) Molecular structures and corresponding atomic numbers of pentafluoropyridine;(b)–(d) geometric structure of the S0 state was calculated under B3LYP,M062X and SA-CASSCF(8,8) methods.
在B3LYP,M062X 和SA-CASSCF(8,8)计算水平下,S1态和S2态的几何结构都是明显地具有芳香环平面外畸变的非平面构型.S1态和S2态的几何结构分别如图2 所示,具体的结构参数列在表1 中.在B3LYP,M062X 和SA-CASSCF(8,8)计算方法下得到的S1态的几何结构相似的,都是具有Cs对称性的半船型结构.S1态的C3—F3的键长略长于其他C—F键,而且S1态的芳香环骨架与S0态的相似的,键长没有发生太大变化.在SA-CASSCF(8,8)方法下计算得到的二面角C3—C2—C1—C4,F3—C3—C4—C1,F4—C4—C5—C1和F5—C5—C4—C2比B3LYP,M062X 计算方法得到的结果略低.值得注意的是,在RISCS-CC2 方法下计算的结果也表明S1态的结构是具有明显的平面外畸变的非平面结构,且二面角C3—C2—C1—C4和F3—C3—C4—C1分别为2.5°和35°[22],这些数值与利用SA-CASSCF(8,8)方法得到的结果较为接近.

表1 利用B3LYP,M062X,SA-CASSCF(8,8)方法,得到五氟吡啶分子的S1 态和S2 态的结构参数(键长单位Å,二面角单位(°))Table 1.Structural parameters of the S1 and S2 states were obtained by B3LYP,M062X and SA-CASSCF(8,8) methods,respectively (Bond length and dihedral angle are Å,(°) in units,respectively).
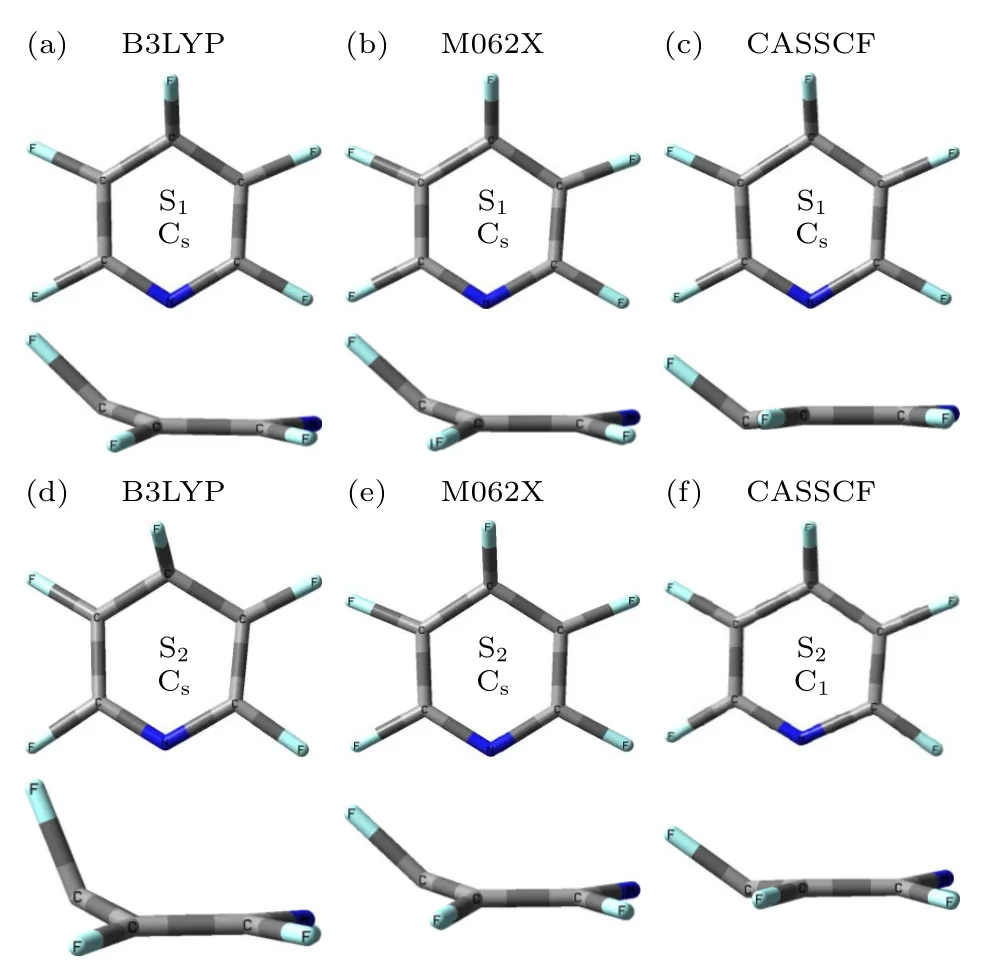
图2 利用B3LYP,M062X 和SA-CASSCF(8,8)方法优化得到S1 态(上图)和S2(下图)的几何结构Fig.2.Geometric structures of the S1 (upper) and S2(lower) states were calculated under B3LYP,M062X and SA-CASSCF(8,8) methods.
同样,在B3LYP,M062X 和SA-CASSCF(8,8)计算方法下,S2态也是具有平面外弯曲的半船型结构.利用SA-CASSCF(8,8)方法计算得到的几何结构和B3LYP,M062X 方法得到的结果略有不同.利用SA-CASSCF(8,8)方法计算得到S2态的构型具有C1对称性,而DFT 方法下B3LYP 和M062X 给出的结构具有Cs对称性.在B3LYP和M062X 计算水平下,结果显示S2态的C3—F3的键长略长于其他C—F 键的键长,这与S1态的情况一样.而SA-CASSCF(8,8)方法得到的S2态的C3—F3的键长是在所有C—F 键中最短的,而且两个C—N 键的键长不再一致,这也导致了对称性的降低.利用B3LYP 方法得到的二面角F3—C3—C4—C1是75.49°,远大于其他两种方法的结果.有趣的是,在SA-CASSCF(8,8)方法下,二面角F4—C4—C5—C1和F5—C5—C4—C2几乎是在B3LYP,M062X 方法下所获得的结果的两倍.同样地,在SA-CASSCF(8,8)方法下,二面角N—C1—C2—C5是20.69°,远大于B3LYP 和M062X方法的结果.通过比较吡啶、六氟苯和五氟吡啶分子的几何结构之间的差异[21,22,29–32],表明具有较高对称性的芳香族分子的激发态通常具有非平面结构.
3.2 吸收光谱
利用B3LYP,SA-CASSCF(8,8),M062X,CASPT2 等方法分别计算了五氟吡啶分子S1态和S2态的VEEs 和AEEs,获得的能量列在表2中.由表2 可以看出,由于计算对S1的绝热激发能做了零点能修正,相对于垂直激发能,绝热激发能与实验值的偏差相较小,仅在4%左右.B3LYP 方法计算得到的吸收谱中两个吸收带的中心分别位于5.33 eV 和6.28 eV,而M062X 方法得到的两个吸收带的中心分别位于5.63 eV 和6.50 eV.此外,利用SA-CASSCF(8,8)计算方法,S1态的VEE和AEE 分别为5.47 eV 和4.84 eV,S2态的VEE和AEE 分别为6.92 eV 和6.69 eV.利用CASPT2方法获得的激发态的能量小于其他方法所得到的能量,S1态的VEE 和AEE 分别为5.02 eV 和4.41 eV,S2态的VEE 为6.33 eV.此外,由于S2的激发能缺少实验数据,表2 中没有给出VEE 和AEE 的偏差.图3 给出了B3LYP 和M062X 方法下获得的五氟吡啶分子的吸收光谱,虚线代表计算不同方法下的各个能量位置,吸收谱使用高斯函数展宽,展宽参数为0.5.从计算的吸收光谱可以观察到,五氟吡啶分子在紫外波长范围内存在两个较宽的吸收带,计算得到的吸收光谱与实验结果[22]相吻合.相对于M062X 方法获得的吸收谱,B3LYP方法获得的吸收谱向长波方向偏移.
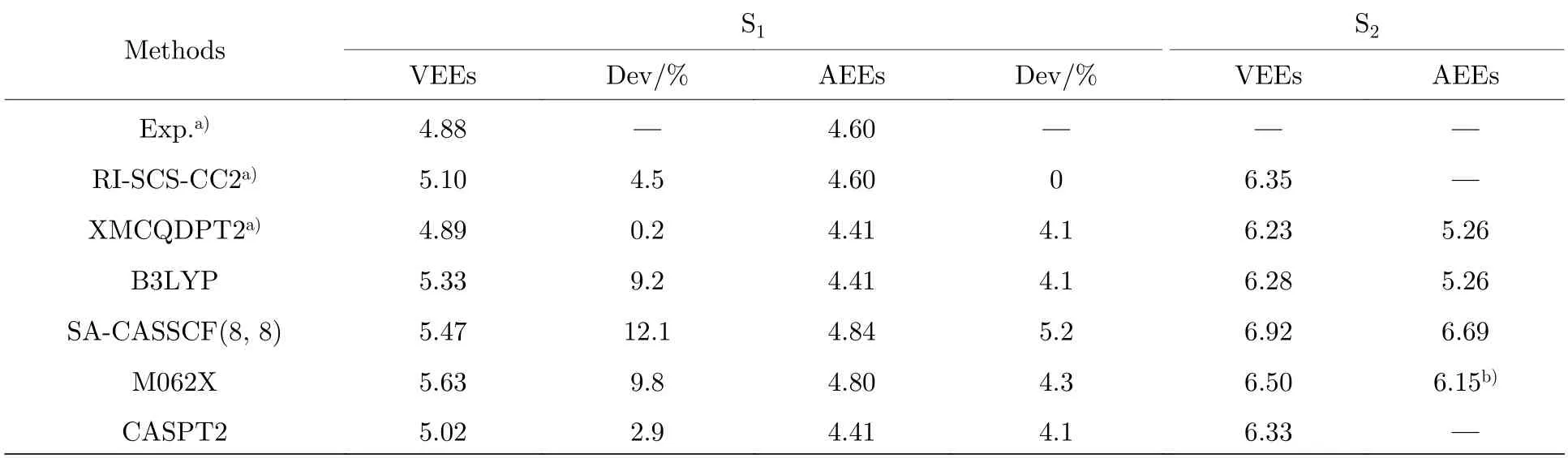
表2 B3LYP,SA-CASSCF(8,8),M062X 和CASPT2 方法结合6-311G*基组计算得到五氟吡啶分子S1 态和S2 态的VEEs 和AEEs (单位为eV)Table 2.VEEs and AEEs (in eV) of pentafluoropyridine in the S1 and S2 states calculated at B3LYP,SA-CASSCF(8,8),M062X and CASPT2 levels with the 6-311G* basis set.

图3 利用B3LYP 和M062X 方法计算得到的五氟吡啶分子吸收光谱Fig.3.Absorption spectra of pentafluoropyridine by B3LYP and M062X levels.
3.3 锥形交叉结构和性质
利用SA-CASSCF 方法计算了锥形交叉S2/S1,S1/S0和S2/S0的结构,结构如图4 所示,结构的具体参数列在表3 中.计算表明锥形交叉S2/S1,S1/S0和S2/S0的结构都是非平面结构,分别是船型、半船型和椅式结构.在3 个锥形交叉的结构中所有C—F 键的键长相似,键长是1.30—1.32 Å.除锥形交叉S2/S1中的C2—C3键的键长是1.39 Å之外,其余锥形交叉的C—C 键的键长是1.45—1.49 Å.锥形交叉S2/S1结构的C1—N 键和C5—N键的键长相差0.26 Å,分别是1.45 Å和1.29Å,而锥形交叉S1/S0和S2/S0结构的C1—N 键和C5—N键的键长是接近的,仅相差0.2 Å和0.6 Å.锥形交叉S2/S1的结构和本工作中的其他非平面的结构也略有不同.锥形交叉S2/S1的C1,C2,C4和C5四个原子不再在同一个平面内,C1原子和C4原子相对N,C5,C3和C2组成的芳香环平面是平面外弯曲的,且与芳香环平面所形成的二面角C1—C2—C3—N 和C4—C3—C2—N 分别是28.64°和11.80°.此外,和C1原子和C4原子相连的F1原子和F4原子对应的二面角F1—C1—C2—C3和F4—C4—C3—C2分别是35.29°和11.65°.S1/S0的结构是半船型的,只有C3相对C1,C2,C4和C5四个原子所在的芳香环平面是平面外弯曲的,所形成的二面角C3—C2—C1—C4和F3—C3—C4—C1分别是46.24°和36.30°.锥形交叉S2/S0结构中的C3原子和N 原子相对C1,C2,C4,C5四个原子组成芳香环平面向上弯曲,而F1原子和F5原子相对往下弯曲,锥形交叉S2/S0所形成的结构是椅式的.锥形交叉S2/S0结构中的二面角C3—C2—C1—C4和F3—C3—C4—C1分别是12.28°和64.87°,而二面角N—C1—C2—C5以及F1—C1—C2—C4分别是22.59°和60.00°.利用SA-CASSCF 方法计算了锥形交叉S2/S1,S1/S0和S2/S0的能量分别是6.39,5.16 和8.51 eV.相同的计算水平下,S2态的能量仅比锥形交叉S2/S1的能量高0.30 eV,但是远低于锥形交叉S2/S0的能量1.82 eV.这表明处于S2态势能面的波包可以较容易地通过锥形交叉S2/S1弛豫到S1态,然而S2态需要更多的能量才能到达锥形交叉S2/S0,所以通过锥形交叉S2/S0直接弛豫到S0态的过程很难发生.锥形交叉S2/S1的能量比S1/S0的能量大1.23 eV,波包在通过锥形交叉S2/S1后会携带较多的过剩能量,尽管锥形交叉S1/S0的能量比S1态稳定结构的能量高出0.32 eV,这些携带较高的能量的波包弛豫到S1态最小能量点后,很容易再通过锥形交叉S1/S0弛豫衰减到S0态.

表3 SA-CASSCF 水平下的锥形交叉的结构参数(键长单位Å,二面角单位 (°))Table 3.Structural parameters of conical intersections were obtained by SA-CASSCF(8,8) methods(Bond length and dihedral angle are Å,(°) in units).

图4 SA-CASSCF 水平下的锥形交叉的结构(a) S2/S1;(b) S1/S0;(c) S2/S0Fig.4.Structures of conical intersections under the SACASSCF level: (a) S2/S1;(b) S1/S0;(c) S2/S0.
此外,根据文献[33–36]中的公式:
提取了分支空间中的S2/S1,S1/S0和S2/S0的4 个拓扑参数σx,σy,∆gh和dgh,其中σx和σy代表锥形交叉偏离垂直方向的斜率,∆gh和dgh分别描述锥形交叉的对称性偏差程度和倾斜率[33].这4 个拓扑参数列在表4 中.由4 个拓扑参数描绘地锥形交叉S2/S1,S1/S0和S2/S0在分支空间中的结构如图5 所示.3 个锥形交叉S2/S1,S1/S0和S2/S0的σx和σy的值比较小,都具有尖峰且不对称的结构.由于锥形交叉S2/S1和S1/S0的σx和σy值相差较小,S2/S1和S1/S0分别在y方向和x方向略有倾斜.而S2/S0的σx比σy值大0.40 eV/Å,在分支空间中明显地观察到S2/S0向x方向倾斜.参数dgh表明,经锥形交叉S2/S1和S1/S0所形成的锥形交叉面更加陡,而经S2/S0的锥形交叉面相对平坦,呈不对称的尖峰形式.上述结果表明,从S2态直接非绝热转移到S0态的效率比较低.
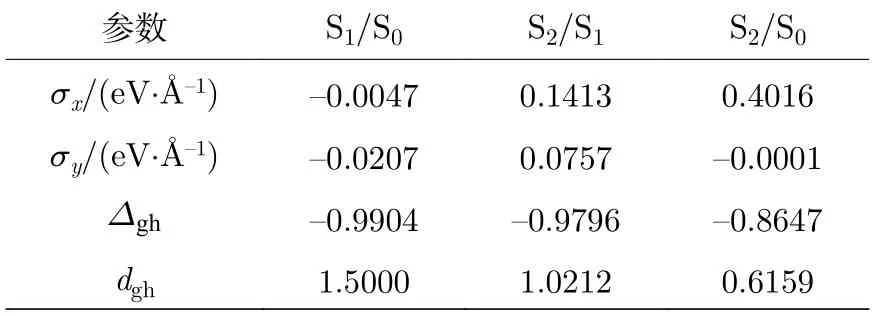
表4 锥形交叉在分支空间中的拓扑参数Table 4.Topological parameters of conical intersections in branching space.
3.4 激发态的弛豫机制
通过分析不同电子态之间的锥形交叉结构和能量,进一步阐明了五氟吡啶分子在激发后所经历的弛豫过程,如图6 所示.通过NEB 方法基于B3LYP/6-311G*水平计算了势能面上连接S2态和锥形交叉S2/S1之间的最小能量路径.B3LYP/6-311G*方法结果表明S2态和锥形交叉S2/S1的能量分别为5.71 eV 和5.41 eV,其能差为0.30 eV,这与CASSCF 方法计算得到的能差0.30 eV 一致.同时NEB 路径表明,在势能面演化过程中存在着0.06 eV 的较小势垒.综合先前的分析可知,五氟吡啶分子被激发到S2态后,波包会在势能面上演化.由于S2态稳定结构下的能量略高于锥形交叉S2/S1的能量,这个演化是一个几乎无势垒的过程,很容易发生.最终波包通过锥形交叉S2/S1快速弛豫到S1态.另外,五氟吡啶分子的锥形交叉S2/S1和S1/S0的能差较大,计算得到的差值是1.23 eV.先前研究表明,苯分子激发态弛豫过程所经历的两个锥形交叉S2/S1和S1/S0的结构相似,被激发到S2态后波包可以在50 fs 内快速地连续通过两个锥形交叉S2/S1和S1/S0直接弛豫到S0态[2,37,38].而五氟吡啶分子在激发态上演化过程中涉及的这两个锥形交叉的结构分别是船型和半船型,具有较大的差别,在演化过程中会发生核结构的变化,这说明五氟吡啶分子的S2态弛豫路径与苯不同,且寿命大于苯的S2态的寿命.此外,飞秒瞬态吸收光谱实验结果表明[31],吡啶被激发到S2态后将在>10 ps 的时间 尺度上 弛豫到S1态,随后以9—23 ps 的时间衰减到基态.同时,S2态弛豫还存在经中间体结构以>2 ns 的时间尺度直接弛豫到基态的路径.由于氟原子取代后的五氟吡啶的激发态结构发生了较大的面外畸变,N 原子相对的C3和F3原子翘起形成了半船型结构,且锥形交叉S2/S1的能量比S1态稳定结构的能量高出1.55 eV,且S1态稳定结构和锥形交叉S1/S0的能差相对较小,这导致五氟吡啶分子在通过锥形交叉S2/S1到达S1态时仍携带了大量的过剩能量,波包先演化到其稳定结构,还可继续再次通过锥形交叉S1/S0弛豫到S0态.此外,当五氟吡啶分子被激发到S1态后,处于S1态的波包将通过锥形交叉S1/S0直接衰减到S0态.先前CASSCF(8,7)结合6-31G(d)或STO-3G 基组的计算结果[29,31]证实,吡啶的锥形交叉S1/S0的结构为半船型结构,其中6-31G(d)基组下结构是与N 原子相邻的C 原子翘起,且能量低于S1态1.56 kcal/mol,而STO-3G 基组下结构为N 原子偏离杂环平面.如前所述,我们计算得到五氟吡啶的锥形交叉S1/S0是N 原子相对的C3原子翘起的半船型结构.CASSCF(8,7)/6-31G(d)的理论结果表明吡啶的S1态衰变是通过锥形交叉S1/S0弛豫至S0态的过程[29].参照先前研究分别得到的苯和吡啶中6.7 ps 和9—23 ps 的S1态寿命[2,22,30,37,39–43],五氟吡啶分子S1态的寿命估计为ps 量级.值得一提的是,锥形交叉S2/S0的存在可能会导致S2态的波包会直接弛豫到S0态,但是锥形交叉S2/S0的能量要远大于S2态稳定结构的能量,结合分支空间中锥形交叉的拓扑结构可以推断出五氟吡啶分子通过S2/S0直接从S2态弛豫到S0态的过程很难发生.

图6 五氟吡啶分子的非辐射弛豫动力学Fig.6.Nonradiative relaxation dynamics of pentafluoropyridine.
4 结论
本文利 用B3LYP,M062X,SA-CASSCF(8,8)和CASPT2 等方法结合6-311G*基组研究了五氟吡啶分子的激发态的结构和能量以及涉及锥形交叉的非辐射弛豫动力学过程.五氟吡啶分子的S0态几何结构是具有C2v对称性的平面结构,而激发态S1态和S2态的几何结构都是具有较强的平面外畸变的半船型.此外,锥形交叉S2/S1,S1/S0和S2/S0的拓扑结构分别确定为船型、半船型和椅式结构.同时,计算了这些结构相应的能量,这些能量值与实验值测量值相吻合.锥形交叉S1/S0,S2/S1和S2/S0的能量分别估计为5.16,6.39和8.51 eV.根据分子的稳定结构和能量以及分支空间中锥形交叉的拓扑结构,分析了激发态演化涉及到锥形交叉的非辐射弛豫途径.五氟吡啶分子被激发到S2态后,波包主要通过锥形交叉S2/S1快速演化到S1态势能面的最小能量处,再通过锥形交叉S1/S0直接衰减到S0态.而S2态的波包直接通过S2/S0弛豫到基态的途径发生的概率较小.
