主要组织相容性复合物Ⅱ类分子与绝经后骨质疏松症的相关性
2024-03-19张桢陈昊王雪鹏朱六龙
张桢 陈昊 王雪鹏 朱六龙
浙江大学医学院附属杭州市第一人民医院骨科,浙江 杭州 310006
1 绝经后骨质疏松与MHC-Ⅱ的概念
绝经后骨质疏松症(postmenopausal osteoporosis,PMOP)是一种主要由雌激素降低引起的骨骼疾病,影响全球数千万女性,该病的主要特征为骨强度和骨密度的降低[1]。相较于同龄男性,女性在绝经岁后往往更容易因骨质疏松导致骨折[2]。既往研究往往聚焦于雌激素降低引起的成骨细胞或破骨细胞的相互作用和活跃程度变化、骨髓间充质干细胞的成骨与成脂分化失衡,然而骨免疫失衡这一概念逐渐被提起。骨免疫失衡主要是指免疫系统和骨骼系统之间的复杂串扰变化诱发的骨稳态失衡。骨稳态指成骨细胞主导的骨形成和破骨细胞主导的骨吸收之间的平衡,骨稳态的失衡往往会导致骨量的改变,从而诱发骨质疏松症[3]。
主要的组织相容性复合体Ⅱ类分子(major histocompatibility complex class Ⅱ molecules,MHC-Ⅱ)由α链和β链组成,它们均含有有外显区,其中α1和β1结构域具有参与抗原结合的功能。MHC-Ⅱ类分子主要在单核巨噬细胞、树突状细胞等抗原呈递细胞上表达,可以发挥一种类似“告示牌”的作用并通过蛋白质递呈途径参与由T和B淋巴细胞介导的适应性免疫反应,从而发挥关键作用[4-5]。值得一提的是,MHC-Ⅱ在破骨细胞前体中高表达,而MHC-Ⅱ类反式激活剂(MHC class Ⅱ trans activators,CIITA)过表达小鼠模型可诱导严重的自发性骨质疏松症[6]。因此,进一步探索MHC-Ⅱ在绝经后骨质疏松症调节骨稳态的潜在机理可能有助于绝经后骨质疏松症靶向药物的开发。
2 MHC-Ⅱ介导T和B淋巴细胞的活化作用
由MHC-Ⅱ介导的T细胞相关的适应性免疫反应和炎症是一个复杂的病理过程。在抗原提呈细胞吞噬例如细菌、蛋白质等细胞外蛋白后,这些蛋白可被吞噬体包裹在细胞中。之后细胞内溶酶体将与吞噬体结合形成吞噬溶酶体,将蛋白质水解成短肽。MHC-Ⅱ分子可以监测巨噬细胞中不同蛋白的水解机制,并与经消化水解后的短肽结合,以MHC-抗原复合物形式转载至抗原提呈细胞表面,从而参与识别和结合初始T细胞和T细胞记忆细胞[7]。T细胞上的抗原特异性T细胞受体与抗原提呈细胞上的抗原肽MHC-Ⅱ的相互作用是T细胞活化的必要阶段[8]。当MHC-抗原复合物与T细胞受体结合后,可以通过钙离子信号、磷脂酰肌醇信号和酪氨酸激酶信号等触发一系列信号传递,从而激活T细胞,并诱导其分化为辅助性T细胞 (T helper 1,Th1)、Th2、Th17等不同亚型[9]。Th细胞又称CD4+T细胞,是组成性表达T细胞受体(constitutive expression of T cell receptors,TCR)以及CD4分子的T细胞,其可在B细胞的活化过程中发挥双重作用。Th细胞首先可通过在表面表达CD40 L,与B细胞表面的CD40结合促进B细胞活化[10]。此外,Th细胞产生一些细胞因子,如白细胞介素-2(interleukin-2,IL-2)、IL-6等可进一步促进B细胞活化。活化的T和B淋巴细胞可产生干扰素-γ(IFN-γ)、肿瘤坏死因子-α(TNF-α)、白细胞介素-17A(IL-17A)和核因子-κB(NF-κB)配体(RANKL)的受体激活剂等免疫分子[11]。这些炎症因子可通过调节破骨细胞在绝经后骨质疏松症骨重建过程中发挥了重要作用。由此可见,MHC-Ⅱ分子递呈途径在T细胞和B细胞激活过程以及随后的骨重建调节过程的多个阶段发挥着起始作用。
3 效应T淋巴细胞和B细胞调控炎症因子介导的骨重建
骨重建是一个复杂的过程,主要包括由破骨细胞主导的破骨作用(用于清除受损的骨组织)和成骨细胞主导成骨作用(用于骨形成)[12]。众所周知,经MHC-Ⅱ的活化后T淋巴细胞和B淋巴细胞往往可以通过分泌各种炎症因子发挥调节体内免疫系统的作用,包括RANKL、TNF-α、IL-17A和IL-17F以及IFN-γ。以上炎症因子则具有调节破骨细胞和成骨细胞的作用,从而发挥调控骨代谢和骨重建的功能。
RANKL已被确定为炎症性骨质流失的关键介质和骨免疫学的关键分子之一。活化的T细胞可以直接产生RANKL,RANKL可进一步结合单核细胞上的RANK,并通过细胞内NF-κB信号传导和激活蛋白(AP)-1转录因子家族诱导破骨细胞形成[13]。此外,激活的RANK可促进肿瘤坏死因子受体相关因子(tumor necrosis factor receptor-related factors,TRAFs)的表达,从而导致破骨细胞分化[14]。TNF-α在骨代谢中也发挥着重要作用,其可通过直接作用于破骨细胞前体,以及间接调控间质细胞产生M-CSF和RANKL来影响破骨细胞的形成[15]。既往研究表明TNF-α还可以通过激活NF-κB和PI3K/Akt信号通路促进RANKL诱导的破骨细胞形成并且可诱导成骨细胞自噬和凋亡,从而破坏骨稳态[16]。IL-17A 是一种主要由Th17细胞分泌的一种免疫细胞因子,参与骨重塑的调节和骨质的破坏。TH17细胞可通过产生IL-17F促进RANKL等破骨分子的作用,对破骨细胞的分化产生间接刺激作用[17]。IFN-γ也称为免疫干扰素,在宿主防御中具有重要作用,对破骨细胞的调节作用具有两面性。其可基于对RANKL信号的干扰,产生抑制破骨细胞分化的效果[18]。IFN-γ还可以刺激破骨细胞产生超氧化物,导致破骨细胞凋亡增强。然而,富含IFN-γ的微环境有利于促进成熟的单核细胞/巨噬细胞融合分化为破骨细胞[19]。
4 雌激素调控MHC-Ⅱ介导的T淋巴细胞活化
雌二醇被证明具有抑制抗原呈递和共刺激分子表达的作用,并且卵巢切除导致的雌激素缺乏可诱导MHC-Ⅱ的异常表达以及抗原呈递增强[20-21]。经免疫荧光染色证明,在OVX小鼠的上皮导管细胞中,MHC-Ⅱ阳性细胞率比对照组增长了近50%。另一项研究表明,17β-雌二醇可以降低小胶质细胞标志物MHC-Ⅱ的表达水平,并实现促进髓鞘再生的作用[22]。树突状细胞(dendritic cell,DC)是骨髓中一种MHC-Ⅱ类分子高表达的抗原呈递细胞,被认为是诱发骨质疏松的潜在因素。大豆黄素因其结构与17β-雌二醇相似,也具有雌激素类似作用。实验证明其具有抑制树突状细胞的功能成熟以及抑制树突状细胞对T细胞的同种异体刺激能力的作用[23]。低水平的雌激素可导致树突状细胞拥有更长的存活时间,这将进一步增加其在抗原呈递途径过程中发挥的作用[24]。因此,雌激素可能具有通过调控MHC-Ⅱ介导抗原呈递途径从而发挥调节骨质疏松的作用。
绝经后妇女伴随着雌激素的降低,可引起骨微环境发生炎症性改变,从而加速骨质的流失以及增加发生骨质疏松症的风险。在此过程中,雌激素的缺乏可导致CD4+T细胞失调,从而诱导微环境内炎症细胞因子循环水平升高,其中RANKL、IFN-γ、IL-17、TNFα和CD40 L增加尤为明显[25-26]。此外,雌激素缺乏可增加活化T细胞上表达的共刺激因子CD40 L数量,诱导基质细胞上M-CSF和RANKL过表达,下调骨保护素(OPG)的产生,最终导致破骨细胞数量显著增加[27-28]。雌激素缺乏刺激还可诱导Th17细胞分化,增加促破骨细胞因子,如TNF-a、IL-6和RANKL的表达水平,最终导致骨质流失[29]。口服补充雌激素可逆转促破骨细胞因子的变化,并发挥抑制Th17分化和IL-17产生的作用,从而保护骨骼健康[30]。T细胞缺陷可使患有卵巢切除术(ovariectomy,OVX)的小鼠中骨质流失减少的症状得到明显改善[31]。以上结果表明雌激素可能具有通过调控T淋巴细胞功能,从而具有调节绝经后骨质疏松症的作用。总之,在绝经后的妇女中,雌激素缺乏可以通过调控MHC-Ⅱ介导抗原呈递途径诱导T淋巴细胞功能失调,从而导致T细胞衍生的炎症因子分泌紊乱,最终通过促进骨吸收等方式导致骨质流失和骨质疏松症。
5 MHC-Ⅱ与绝经后骨质疏松
靶向调控MHC-Ⅱ类分子被证明具有调节破骨细胞的活性的作用,并与骨质疏松的发生发展密切相关。MHC-Ⅱ类转激活因子(MHC class Ⅱ transactivator,CIITA)是一种专门用于抗原呈递的主要转录共激活因子,是MHC-Ⅱ的主要调节因子。MHC-Ⅱ和CIITA 在破骨细胞前体中表达,并在骨质疏松小鼠中的表达水平明显增强[6]。CIITA过表达小鼠模型可通过促进破骨细胞数量增加和体内骨吸收作用增强诱导严重的自发性骨质疏松症。显微CT扫描结果表明,在24周龄时,CIITA过表达小鼠的胫骨骨小梁数量和骨体积分数相较于野生型小鼠减少了约2/3。此外,在OVX小鼠中,IFN-γ水平升高,而其对CIITA的调节具有决定性作用[32]。CIITA的激活可进一步诱导MHC-Ⅱ分子的表达,并促进了活化的T细胞增殖[33]。另一方面,跨膜蛋白173(Tmem173)作为一种MHC-Ⅱ抑制剂,对破骨细胞特异性基因的表达以及细胞分化产生明显的抑制作用[34]。众所周知,抗酒石酸酸性磷酸酶(tartrate-resistant acid phosphatase,TRAP)是破骨细胞中重要的组织化学标志酶,并且在破骨细胞骨吸收中起重要作用。对经RANKL诱导RAW 264.7细胞同时进行Tmem173过表达处理,转染的细胞中TRAP活性相较于对照载体转染细胞相比下调达60.4%,而在骨片上产生的吸收坑面积下降了超过70%。作为一种MHC-Ⅱ类分子高表达的抗原呈递细胞,树突状细胞被发现在T淋巴细胞转化为效应T细胞的过程中发挥调节作用,并促进例如TNF-a和IL-17等炎症因子的产生[24]。当此调节过程被限制后,小鼠体内骨质流失将得到明显改善[35]。以上结果表明MHC-Ⅱ类分子可能在绝经后骨质疏松发挥一定作用。尽管MHC-Ⅱ分子与破骨细胞和骨质疏松的直接联系的实验性研究尚未进行,但有研究通过生物信息学进行了初步的探索[36]。其表明MHC-Ⅱ分子在绝经后骨质疏松富集,并推测可能具有通过直接调节T细胞分泌炎性细胞因子产生调控骨稳态的功能。
然而,有研究表明MHC-Ⅱ的缺失却会对小鼠外周骨骼的发育产生不利影响。与野生型小鼠相比,MHC-Ⅱ敲除小鼠在多个年龄阶段中,其股骨力学、几何和成分测量值等指标明显降低,包括了股骨刚度、3-pt弯曲峰值力和矿物质质量[37]。在小鼠16周龄时,MHC-Ⅱ敲除小鼠的这3项指标分别仅仅为野生型小鼠的80%、66%和61%,并且伴随着体重的降低。尽管其中的潜在机制并未进一步探索,但可能与MHC-Ⅱ在机体内广泛的作用机制有关。因此,MHC-Ⅱ与破骨细胞和骨质疏松的内在联系和潜在机制需要进一步探索。
6 结论
本文聚焦于MHC-Ⅱ分子在绝经后骨质疏松的潜在联系进行综述。雌激素缺乏可促进MHC-Ⅱ分子蛋白递呈途径,进而加剧T淋巴细胞和B淋巴细胞分泌炎症因子,最终导致骨稳态失衡(图1)。尽管本文为揭示绝经后骨质疏松发生的病理机制提供了潜在的治疗的方向。然而,MHC-Ⅱ分子蛋白递呈途径在绝经后骨质疏松的具体作用机制仍需进一步探索。
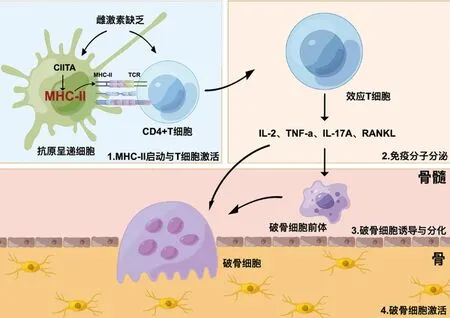
图1 雌激素缺乏靶向MHC-Ⅱ分子调控的骨质疏松症Fig.1 Estrogen deficiency targets major histocompatibility complex Class II molecular-mediated osteoporosis
