Nrf2调控网络与骨质疏松症的相关性及中医药干预研究
2024-03-19刘沛蒋宜伟周玉英张夏阳海云翔旷武龙江朔轩
刘沛 蒋宜伟,* 周玉英 张夏阳 海云翔 旷武龙 江朔轩
1 甘肃中医药大学,甘肃 兰州 730000
2 甘肃中医药大学附属医院,甘肃 兰州 730000
骨质疏松症(osteoporosis,OP)被称为“21世纪的无声流行病”,以骨量降低、骨组织微观结构恶化、骨折易感性增加为主要特点[1]。研究调查显示,中国中老年男性OP患病率为20.73%,女性为38.05%[2],OP已经成为日益严重的健康问题。OP的发病机制错综复杂,与氧化应激、自噬、炎症以及代谢异常有关[3]。核因子E2 相关因子 2(nuclear factor erythroid 2-related factor 2,Nrf2)是一种主要通过调节细胞氧化还原稳态发挥细胞保护作用的内源性转录因子。此外,Nrf2还参与许多其他细胞过程,包括调节炎症、自噬、铁死亡等多个方面[4]。OP的发生主要是由于骨稳态的破坏,导致成骨细胞介导的骨形成与破骨细胞的骨吸收之间平衡被打破,使骨吸收大于骨形成从而导致OP,而氧化应激、自噬、炎症等均会导致或者加剧这一过程。Nrf2通过复杂的调控网络能够有效调节上述因素改善OP,且中医药以Nrf2为靶点在治疗OP上优势显著、效果明显,因此笔者将从中医药通过Nrf2相关调控网络治疗骨质疏松症的研究进展做一综述。
1 Nrf2概述
Nrf2是直接与抗氧化反应元件(ARE)直接结合的主要核转录因子,其具有七个保守的Nrf2-ECH同源(NEH)结构域,它们具有不同的功能来控制Nrf2的转录活性[5]。Kelch样ECH相关蛋白1 (Keap1)是Nrf2的主要胞内调控因子,在正常稳态条件下,Nrf2被Keap1隔离在细胞质内,在氧化应激的情况下,Keap1蛋白质构象发生改变,Nrf2/keap1之间相互作用被破坏,Nrf2游离入细胞核,结合s Maf蛋白后,Nrf2激活抗氧化反应元件部位(ARE)增加其调节下游抗氧化基因的表达[6],血红素加氧酶-1(HO-1)是血红素分解代谢和应激诱导的细胞保护酶,主要通过Nrf2调节,Nrf2的激活可以刺激HO-1的表达,HO-1则通过其代谢产物发挥抗氧化的作用[7]。NADP (H):醌氧化还原酶-1 (NQO-1)作为一种黄素酶,同样是维持细胞氧化还原状态的重要抗氧化酶[8],在Nrf2被激活的同时刺激NQO-1的表达,NQO-1通过与蛋白质的相互作用以及mRNA翻译的结合和调节等通过多个方面抗氧化应激[9]。
激酶磷酸化对Nrf2的活化以及下游靶点的调节是至关重要的,糖原合成酶激酶3β(GSK-3β)可直接磷酸化Nrf2,从而抑制Nrf2靶基因的表达,此外,GSK-3β磷酸化Src家族酪氨酸激酶Fyn以促进其核积累,通过增加Nrf2的核输出和降解来负调控Nrf2活性[10]。作为Nrf2上游信号的磷脂酰肌醇3(PI3K)/蛋白激酶B(Akt)通过阻断GSK-3β活性,增加Nrf2水平并激活下游抗氧化途径[11]。
2 Nrf2与OP
Nrf2是一种在许多细胞类型中表达的转录因子,包括成骨细胞、破骨细胞和骨细胞,Nrf2在骨稳态的调节中起着重要的作用。研究发现,Nrf2的缺乏促进了OP的进展,并且与增龄性氧化应激相关[12]。在成骨细胞到骨细胞的过渡过程中,由于线粒体含量增加导致活性氧(reactive oxygen species,ROS)水平增加,而作为氧化应激的主要传感器,Nrf2在骨细胞生成过程中被激活,并直接反式激活许多骨细胞特异性基因,如Dmp1、Mepe和Sost。并且在动物实验研究中发现,Nrf2缺失的小鼠导致了骨质的减少并损害骨细胞特异性基因的表达[13]。此外Nrf2的激活是抑制骨吸收的关键,一方面其可以通过减少ROS的产生直接抑制破骨细胞的分化,另一方面激活Nrf2/ARE信号通路可以通过控制细胞内ROS信号传导来调节破骨细胞的分化[14]。HO-1作为Nrf2下游的靶点,对于破骨细胞的分化同样具有负向调节作用,Nrf2/HO-1途径通过抑制NF-κB信号的传导来抑制NF-κB受体激活配体(RANKL)诱导的破骨细胞分化[15]。Nrf2对成骨细胞的影响主要体现在保护其免受氧化损伤方面。研究发现,增加 GSH/GSSG比率可以通过 PI3K/Akt-Nrf2信号通路减弱ROS对成骨细胞的氧化损伤[16],在MC3T3-E1细胞中miR-455-3p的过表达通过负向调控组蛋白去乙酰酶(histone deacetylases,HDACs),并激活Nrf2/ARE途径促进成骨细胞增殖分化,并且减轻了OP小鼠氧化应激损伤[17]。由此可见,Nrf2通过多种途径促进骨形成,抑制骨吸收,在维持骨稳态中扮演着重要角色。
3 Nrf2调控网络与OP的相关性
3.1 Nrf2调控氧化应激
氧化应激是引起OP的重要危险因素,其诱导的DNA损伤、细胞凋亡和细胞衰老是导致骨稳态失衡的重要原因[18]。氧化应激可以增加破骨细胞的活性,减少成骨细胞活性和分化,并增加成骨细胞和骨细胞的凋亡。氧化应激中NF-κB受体激活配体(RANKL)上调和骨保护素(OPG)下调引起破骨细胞的生成增加,成骨细胞活性降低导致OPG生成减少。这种降低进一步改变了总的RANKL/OPG比率,使成骨细胞和破骨细胞活性失衡而导致骨密度的降低[19],在OP当中,机体氧化应激与衰老、雌激素降低密切相关,雌激素的减少导致氧化还原稳态紊乱,使骨骼更容易受到氧化损伤,最终引起氧化应激,增加患OP的风险。研究发现,卵巢切除的大鼠超氧化物歧化物酶(superoxide dismutase,SOD)、谷胱甘肽过氧化物酶(glutathione peroxidase,GPX)、谷胱甘肽(glutathione,GSH)水平明显降低,同时也增加了大鼠血清骨钙素、碱性磷酸酶(alkaline phosphatase,ALP)、丙二醛(malondialdehyde,MDA)水平和RANKL的表达[20]。Nrf2作为调节氧化应激的关键因子,其控制一系列抗氧化反应元件依赖性基因的基础和诱导表达,对于氧化应激的调节涉及多个途径[21]。并且多项研究表明,Nrf2通过调控氧化应激在维持骨稳态方面起着关键作用。
3.2 Nrf2调控炎症
炎症微环境的改变是引起OP的另一重要危险因素。研究发现,在女性OP患者当中,促炎标志物白细胞介素IL-6、IL-1β、肿瘤坏死因子-α(TNF-α)血清水平明显高于对照组[22]。炎症与骨质流失和破骨细胞的形成密切相关,TNF-α可以直接作用于破骨细胞,与RANKL协同作用,促进破骨细胞生成。TNF-α还通过调节成骨相关转录因子RUNX2的表达抑制间充质基质细胞(mesenchymal stromal cell,MSC)向成骨细胞的分化[23],IL-1β则可以通过NF-κB的机制增强TNF-α诱导的破骨细胞的形成[24]。Nrf2通过调节抗炎基因的表达来抑制炎症的进展,在NF-κB被氧化应激激活时,相关促炎因子将会过量产生[25],NF-κB的活化也会抑制成骨细胞的分化[26]。Nrf2的激活可以阻止促炎细胞因子的转录上调,HO-1及其代谢产物在Nrf2介导下具有显著的抗炎作用,可以降低TNF-α和IL-6的水平[27]。研究发现,激活Nrf2介导HO-1表达升高,抑制NF-κB信号通路,能够有效减轻雄性SD大鼠肝移植模型肠黏膜损伤和紧密连接功能障碍[28]。在面对炎症刺激时,Nrf2通过抑制促炎细胞因子的过度生成以及刺激下游抗炎基因的表达并抑制NF-κB的激活有效发挥抗炎作用,调控炎症反应。
3.3 Nrf2调控自噬
自噬是细胞自我更新的方式之一,其将自身的细胞质蛋白质或细胞器吞噬成囊泡,与溶酶体融合形成自噬溶酶体,并降解溶酶体内容物,实现细胞内稳态和细胞更新[29]。自噬与骨髓间充质干细胞、成骨细胞以及破骨细胞的增殖分化、功能发挥密切相关,自噬活性水平的失调会扰乱骨稳态的平衡,从而介导OP的发生发展[30]。研究发现,通过对小鼠删除自噬相关负调节基因RUBCN后,提高了小鼠成骨细胞的分化和矿化能力,以及参与成骨细胞功能的相关关键转录因子的表达也得到增高[31]。Nrf2对自噬的调控是与自噬衔接蛋白p62相互作用实现的,p62的磷酸化增加了其与keap1的亲和力,并且诱导Nrf2靶点的表达[32]。Tang等[33]研究发现敲除Nrf2后,降低了椎间盘变性小鼠自噬相关基因的表达,而且在氧化应激条件下,自噬作为Nrf2/keap1/p62途径激活的一种抗氧化反应,以保护椎间盘免受变性。由此可见,Nrf2可能通过Nrf2/keap1/p62途径诱导自噬,调节细胞稳态。
3.4 Nrf2调控铁死亡
铁死亡(ferroptosis)是一种新型程序性细胞死亡方式,其特点是铁代谢异常和脂质过氧化的紊乱。铁死亡的发生与过量的Fe2+以“芬顿反应”的方式氧化脂质、产生大量的ROS有关[34]。目前,国内外已有大量研究表明铁死亡与OP有着密切的关系。铁过载在一定程度上抑制成骨细胞的活性、增殖及矿化能力,并导致ROS累积,降低了谷胱甘肽过氧化物酶4(GPX4)蛋白的表达,最终诱导成骨细胞铁死亡导致骨稳态失衡而引起OP[35]。Nrf2在调控铁死亡方面同样起到了不可忽视的作用。多种抑制脂质过氧化与铁死亡的蛋白质和酶都是Nrf2的靶基因,例如铁蛋白的轻链(FTL)和重链(FTH1)、铁转运蛋白(SLC1A2)等受到Nrf2调控,此外抑制铁死亡的两个关键靶点GPX4和胱氨酸/谷氨酸反向转运蛋白(xCT)均是Nrf2下游转录靶标[36]。Nrf2可以直接或者间接调节GPX4的表达和功能,而Nrf2/Keap1信号通路的激活上调了xCT促进细胞增殖并抑制铁死亡[37-38]。p62/Keap1/Nrf2途径的激活也能够有效抑制细胞铁死亡,p62介导的Keap1的降解有助Nrf2的活化,之后Nrf2通过激活下游基因NQO-1、HO-1和以及FTH1的转录使细胞免受铁死亡[39]。研究表明,褪黑素能够通过激活Nrf2/HO-1信号通路抑制高糖诱导的成骨细胞铁死亡,提高成骨细胞的功能,并且改善了糖尿病骨质疏松症(diabetic osteoporosis,DOP)大鼠的骨微观结构[40]。综上,Nrf2对于铁死亡的调控是以Nrf2为中心,通过多种途径实现,一方面是直接调控铁死亡的关键标志物GPX4及关键靶点xCT,另一方面通过调控其下游靶基因防止脂质过氧化及铁过载从而抑制铁死亡。
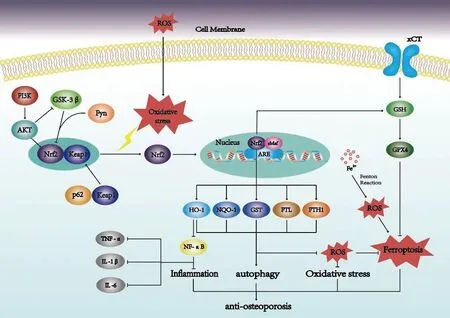
图1 Nrf2调控网络与骨质疏松症Fig.1 Nrf2 regulatory network and osteoporosis
4 中医药通过Nrf2调控网络治疗OP
中国传统医学将OP归为“骨痿、骨痹”等范畴,并且认为OP的发生与肾密切相关,肾虚是OP发病的根本原因[41],肾主骨生髓,肾虚则髓化生无源,无以充养骨骼从而引起OP。近年来研究证实多种补肾类中药单体及其成分、复方以及中成药通过Nrf2途径调控氧化应激、炎症等方面在治疗OP中效果显著。见表1。

表1 中医药通过Nrf2调控网络治疗骨质疏松症的作用机制Table 1 Mechanism of action of traditional Chinese medicine treating osteoporosis through Nrf2 regulatory network
4.1 中药活性成分
多种具有补肾之功的中药单体及其活性成分具有显著的抗OP作用。槲皮素是杜仲中主要类黄酮提取物,研究发现,槲皮素能够通过发挥抗炎、抗氧化应激以及调节细胞凋亡来协调骨代谢,调节骨稳态[42]。淫羊藿被广泛应用于抗炎、抗OP等多种疾病,淫羊藿苷是淫羊藿中主要的黄酮类化合物,Nrf2信号在淫羊藿苷发挥药理作用中起到关键作用[45]。Nrf2是中药活性成分在治疗OP中的关键靶点。
4.2 中药复方
4.2.1左归丸:左归丸具有壮水济火、纯补无泻之功用。研究发现,左归丸提高了过氧化氢诱导的乳鼠颅骨成骨细胞中SOD的含量,降低了MDA含量,并且增加了HO-1和Nrf2蛋白的表达,表明左归丸通过激活Nrf2/HO-1信号通路,降低成骨细胞氧化损伤程度,发挥保护作用[51]。
4.2.2二仙汤:二仙汤由具有温肾阳、补肾精之功效。体内外研究显示,二仙汤增加了TNF-α诱导的成骨细胞中Akt的磷酸化,并且激活了Nrf2、HO-1的表达,减弱了OP大鼠模型中TNF-α的产生,并保护成骨细胞免受TNF-α诱导的细胞凋亡,结果表明二仙汤可能通过调控Akt /Nrf2/HO-1信号通路减轻OP[52]。
4.2.3二仙药对:二仙药对从二仙汤化裁而来,即淫羊藿-仙茅药对,两药相须配伍,补肾强筋之功效增强。在高糖环境下成骨细胞中ROS含量增加,最终发展为DOP,二仙药对通过激活高糖环境下成骨细胞中Nrf2的表达,下调ROS含量,改善高糖成骨细胞氧化应激损伤,从而有效防治DOP[53]。
4.3 中成药
4.3.1正清风痛宁:正清风痛宁的主要成分是青藤碱,青藤碱是中药青藤根茎中的提取物。有研究发现,青藤碱通过调节间充质干细胞中可溶性免疫抑制因子,具有抗炎、抗关节炎、免疫抑制、抑制破骨细胞分化等广泛的药理作用[54]。吴春根等[55]学者通过研究指出,正清风痛宁可以激活Nrf2/SIRT3/SOD2信号通路、抑制氧化应激反应从而促进成骨分化以及抑制破骨分化,提高OP大鼠模型骨密度,改善醋酸泼尼松诱导的骨质疏松。
4.3.2复方鹿茸健骨胶囊:复方鹿茸健骨胶囊具有补肾壮骨、调和气血功效的功效,有显著的抗OP作用[56]。研究表明,复方鹿茸健骨胶囊通过激活Nrf2/HO-1信号通路,抑制过氧化氢诱导的成骨细胞凋亡,维持氧化还原稳态,同时增加成骨相关转录基因RUNX2、Osterix(Osx)等表达,从而有效发挥抗OP作用[57]。
5 总结与展望
OP的发病与机体氧化应激引起的炎症水平升高以及脂质过氧化和铁过载诱导的成骨细胞铁死亡等因素密切相关。Nrf2是一种内源性转录因子,主要作用是保护细胞免受氧化应激反应。Nrf2能够通过一系列途径,激活下游抗氧化基因的表达,在维持细胞氧化还原稳态方面起着关键的作用。Nrf2及其下游靶基因通过调控细胞自噬、抗炎、抑制铁死亡等,是治疗OP中的一个重要的靶点。中医药无论是单体、复方还是中成药通过Nrf2调控网络在治疗OP中效果显著,优势明显,但是中医药通过Nrf2调控铁死亡以及以Nrf2为靶点调节细胞自噬水平在治疗OP方面的研究较少。因此,在未来的研究中应该更加深入中医药通过Nrf2调控网络治疗OP的研究,为中医药治疗OP提供新的方向。
