NFATc1在骨质疏松症中的研究进展
2024-03-19党祎姚红林袁能华马亚萍张怡王信
党祎 姚红林 袁能华 马亚萍 张怡 王信*
1.遵义医科大学附属医院骨科,贵州 遵义 563003
2.遵义医科大学附属医院手术室,贵州 遵义 563003
3.遵义医科大学公共卫生学院,贵州 遵义 563000
骨质疏松症的发生是骨重塑过程不平衡所致,主要是由于破骨细胞对骨的重吸收超过了成骨细胞介导的骨形成[1]。作为破骨细胞分化和破骨细胞基因表达的主要调控因子之一,抑制活化T细胞胞质核因子1(nuclear factor of activated T cell cytoplasmic 1,NFATc1)的表达和易位及其活性可减少破骨细胞的形成和骨吸收,从而保证骨稳态平衡并减少骨疾病的发生[2]。本文拟综述NFATc1影响破骨细胞的生成与分化,及NFATc1与骨修复材料的研究进展,希望能对调控NFATc1靶点治疗骨质疏松症的基础、临床及骨组织工程应用提供帮助。
1 骨质疏松症
骨量减少和骨组织微结构异常是骨质疏松症的显著特征,这一普遍存在的全身性骨病导致骨脆性增强,进而导致骨折风险增加[3]。目前认为,骨质疏松症的重要机制之一在于破骨细胞介导的骨吸收作用超过了成骨细胞介导的骨形成作用,从而导致了骨平衡的破坏[4],见图1。因此,为了达到理想的骨质疏松治疗效果,需要通过一系列复杂而紧密的调控过程来维持破骨细胞的骨吸收和成骨细胞的骨基质形成之间适当的平衡状态[5],从而有效降低骨疾病的发生率。
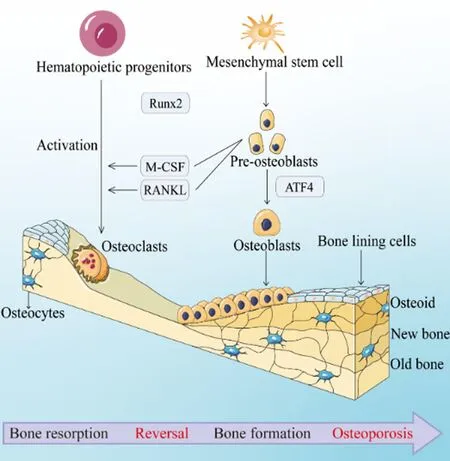
图1 骨质疏松症的发病机制Fig.1 Diagram of the pathogenesis of osteoporosis
2 NFATc1
作为活化T细胞核因子家族(nuclear factor of activated T cell,NFAT)的重要一员,活化T细胞核因子1(nuclear factor of activated T cell cytoplasmic 1,NFATc1)早在20世纪80年代首次被Shaw等[6]在激活的T细胞中发现。NFATc1常驻于T细胞胞质中,其表达受钙调磷酸酶的调控:T细胞受体与抗原的连接触发了受体相关的酪氨酸激酶的激活,从而导致了磷脂酶C-γ(Phospholipase C,PLC-γ)的激活。激活的PLC-γ导致磷脂酰肌醇-4,5-二磷酸的水解,从而产生肌醇-1,4,5-三磷酸(inositol1,4,5-trisphosphate,IP3)和二酰基甘油(diacylglycerol,DAG)。IP3与其受体结合,并向细胞质释放Ca2+离子。钙调蛋白(calmodulin,CaM)捕获游离Ca2+离子并激活磷酸酶钙调磷酸酶,促使NFATc1中的丝氨酸残基去磷酸化,从而激活NFATc1并进行扩增,同时将其易位到细胞核中,进而激活靶基因的转录[7-8]。NFATc1不仅在免疫系统、心脏瓣膜和间隔的形成及动脉粥样硬化中不可或缺,还对于破骨细胞的调节分化至关重要[9]。已有研究证实[10],通过驱动NFATc1的转录控制破骨细胞分化能够调节骨稳态。这一发现不仅揭示了NFATc1调控破骨细胞分化和骨吸收的机制为进一步探索NFATc1对破骨细胞的调节作用尤为必要。
2.1 NFATc1通过相关信号通路介导破骨吸收
破骨细胞源于巨噬细胞/单核细胞系来源的前体细胞,在巨噬细胞集落刺激因子(macrophage colony-stimulating factor,M-CSF)和核因子-κB受体激活剂配体(receptor activator of nuclear factor kappa B ligand,RANKL)的刺激下分化而成,其具备骨吸收功能从而影响骨重塑平衡状态。在破骨细胞生成早期,RANK被触发激活且IP3诱导Ca2+内流,随后激活NFATc1从而促进多种破骨基因如抗酒石酸酸性磷酸酶(tartrate-resistant acid phosphatase,TRAP)及组织蛋白酶K(Cathepsin K,CTSK)等的表达[11-12]。因此,NFATc1在调节破骨细胞相关基因方面发挥着重要的调控作用。Zhang等[13]研究发现转录因子Bhlhe40的缺乏不仅导致体内骨量增加和破骨细胞分化减少,还能够减轻骨质疏松症的发展,而这一过程的发生正是通过直接结合NFATc1和c-Fos的启动子区域并调控其表达所致。另有研究指出,NFATc1缺陷小鼠由于破骨细胞生成受损从而导致发生骨质疏松[14]。这些研究结果表明,NFATc1是破骨细胞分化的重要调控因子。作为破骨分化的重要特异性调节器,NFATc1已被证实常参与RANKL、NF-κB、MAPK及Ca2+等信号通路调控骨吸收过程[15-17],相关信号通路调节过程将在以下进行具体阐述,见图2。
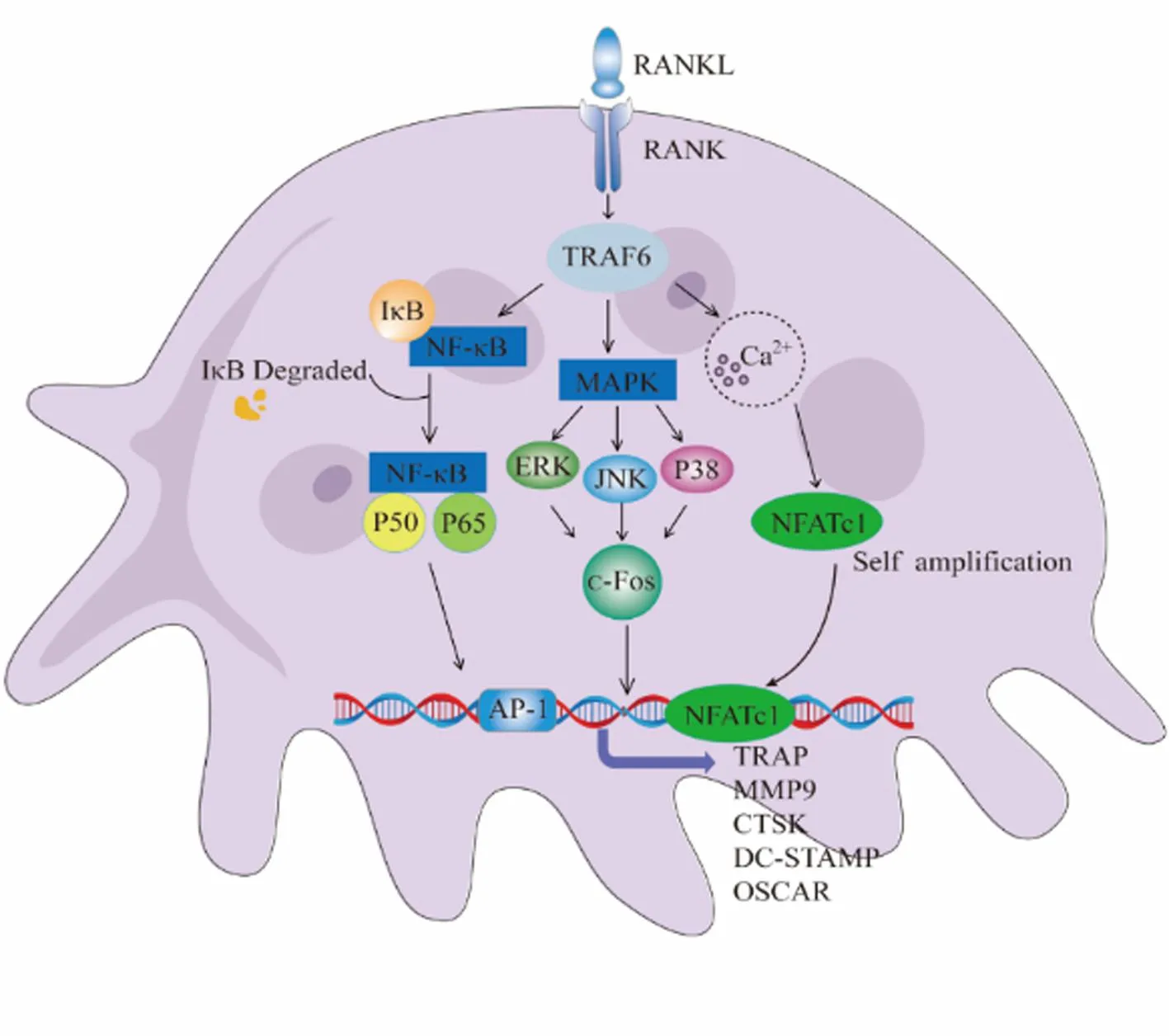
图2 NFATc1参与RANKL、NF-κB、MAPK及Ca2+等多种信号通路介导骨吸收过程的作用机制Fig.2 Mechanism of action of NFATc1 involved in various signaling pathways such as RANKL, NF-κB, MAPK, and Ca2+ to mediate the process of bone resorption
2.1.1RANK-RANKL信号通路:RANKL是一种Ⅱ型跨膜蛋白,其羧基末端具有胞外结构领域。这类细胞外域被基质金属蛋白酶等酶类切割,随后以可溶性形式释放RANKL至细胞外环境中[18]。在早期阶段,RANKL通过与巨噬细胞表面表达的RANK受体结合的一系列中间信号,直接引发破骨细胞从破骨前体细胞向破骨细胞分化[19]。作为破骨细胞中RANK-RANKL信号通路的下游转录因子,NFATc1在调控破骨细胞分化方面扮演着至关重要的角色[20]。具体来讲,RANKL与受体RANK的结合导致衔接蛋白的募集如肿瘤坏死因子受体相关因子6(tumor necrosis factor receptor-associated factor 6,TRAF6),其参与激活下游信号级联反应,如NF-κB、c-Jun、c-Fos和激活蛋白1(activator protein,AP-1)[20],最后诱导以NFATc1为主的转录因子的激活与表达,并促进其去磷酸化和核转位,从而诱导产生破骨分化所必需的基因如TRAP、降钙素受体和CTSK[21],这对于破骨细胞的生成至关重要。Lee等[22]研究发现,通过抑制丙酮酸脱氢酶激酶拮抗RANK-RANKL信号通路能够降低细胞核中NFATc1蛋白的表达水平,从而抑制破骨基因如TRAP、CTSK等的表达,进而阻止破骨细胞分化和骨吸收。这也证实了RANK-RANKL作为上游介导的以NFATc1为靶点的信号通路对破骨形成的影响。此外,RANKL还能够刺激免疫球蛋白样受体如骨髓细胞上表达的触发受体2(triggering receptor expressed on myeloid cells 2,TREM-2)、信号调节蛋白β-1(signal-regulatory protein β-1,SIRP-β1)、唾液酸结合免疫球蛋白样凝集素15(sialic acid-binding immunoglobulin-like lectin 15,Siglec-15)、破骨细胞相关受体(osteoclast-associated receptor,OSCAR)、配对免疫球蛋白样受体A(paired immunoglobulin-like receptor A,PIR-A)和FcγR Ⅲ,这些受体与DNAX-活化蛋白(DNAX-activating protein of 12 kDa,DAP12)和Fc受体γ链(Fc receptor γ-chain,FcRγ)结合介导免疫酪氨酸激活基序(immunoreceptor tyrosine-based activation motif,ITAM)信号传导。ITAM磷酸化导致脾酪氨酸激酶(spleen tyrosine kinase,Syk)的募集,进而激活B-细胞连接蛋白(B cell linker,BLNK)和含SH2结构域的76 kDa的白细胞蛋白(SH2 domain-containing leukocyte protein of 76 kDa,SLP76)等衔接蛋白,这些衔接蛋白作为支架募集布鲁顿酪氨酸激酶/酪氨酸蛋白激酶(bruton tyrosine kinase/tyrosine-protein kinase Tec,Btk/Tec)和PLC-γ,形成破骨细胞信号复合物。该复合物有效激活钙信号,这些信号级联最终导致NFATc1的诱导和激活[23],见图3。综上,NFATc1的生成受到RANK-RANKL信号通路的精准调控,从而对破骨细胞的形成产生了显著影响。
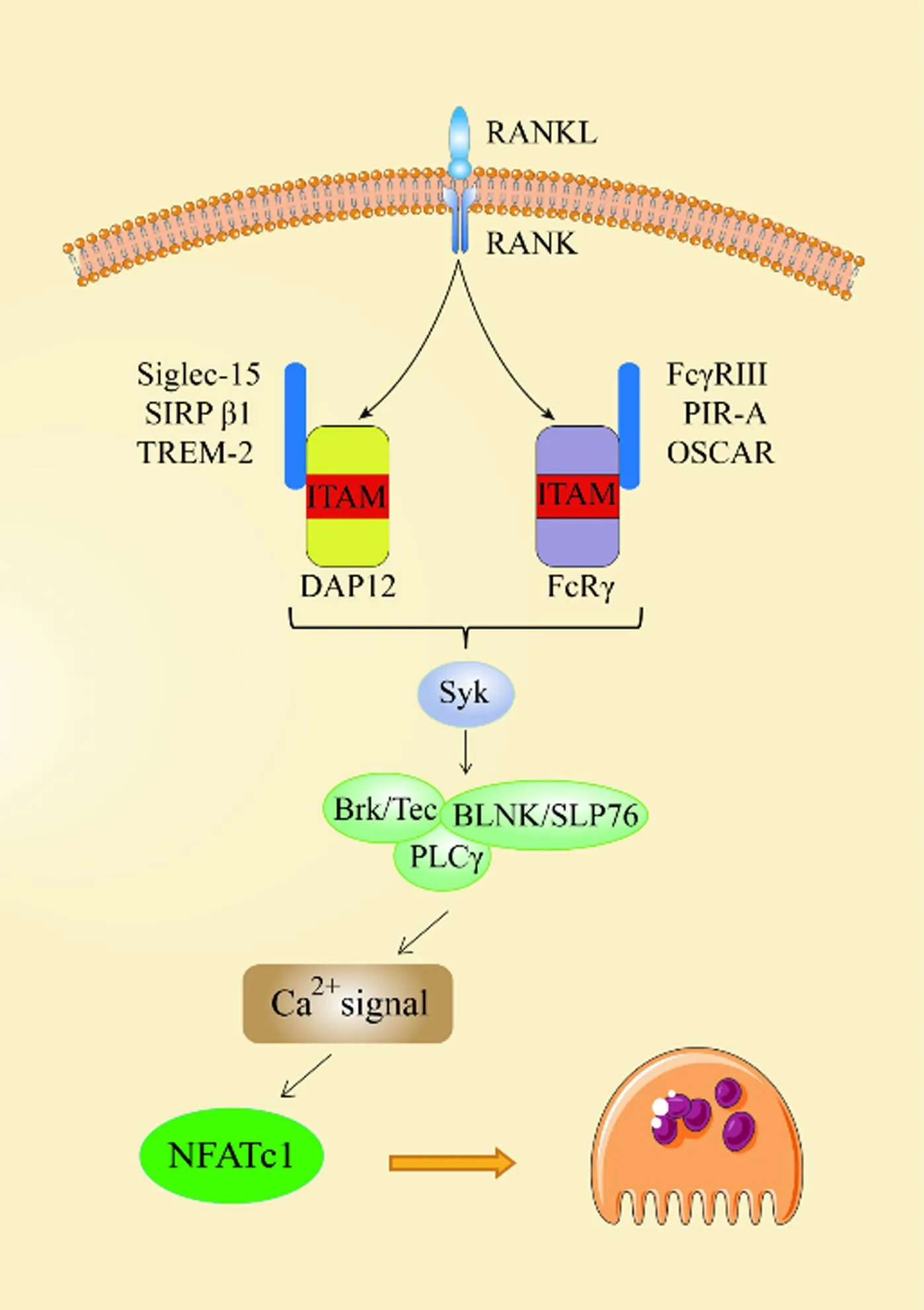
图3 RANKL信号通路在破骨细胞发生中的作用Fig.3 The role of RANKL signaling pathway in osteoclastogenesis
2.1.2NF-κB信号通路:NF-κB作为RANK-RANKL活化的一条主要下游信号通路,在破骨细胞的发生中发挥的作用不可或缺[24]。在典型NF-κB信号途径中,RANKL和RANK的结合可进一步诱导核因子κB抑制因子α(inhibitory Subunit Of NF Kappa B Alpha,IκB-α)降解和p65亚基磷酸化,p65转运到细胞核中启动破骨细胞相关基因和蛋白包括TRAP、c-Fos、CTSK和基质金属蛋白酶9(matrix metalloproteinase-9,MMP-9)的表达[25]。不仅如此,NF-κB的p50和p65亚基响应RANKL,通过与NFATc1启动子区结合诱导NFATc1表达[26]。已有研究发现,NF-κB抑制剂是通过降低NFATc1对RANKL信号通路的抑制作用来诱导破骨细胞的产生[27]。而Liu等[24]的研究也发现麻黄素具有破坏破骨细胞分化和吸收功能,而这一机制主要是通过减弱RANKL刺激的NF-κB信号通路和NFATc1活性来降低破骨基因和蛋白的表达来实现的。由此证明了NF-κB在RANKL-RANK信号诱导NFATc1的激活中的上下游调节关系及其在破骨分化中的调节作用。
2.1.3MAPK信号通路:破骨细胞分化受细胞因子RANKL和M-CSF调控。在破骨细胞分化和骨吸收过程中,M-CSF和RANKL均通过MAPK信号传导发挥作用。M-CSF与其受体的结合导致MAPK和Akt的激活,从而促进破骨细胞的存活[28]。RANKL则与RANK受体结合可募集TRAF6,从而启动下游信号通路MAPK,并激活其下游NFATc1的转录进而调控破骨分化[29]。因此,MAPK信号通路在破骨细胞分化过程的重要性不言而喻。MAPK是一种包含细胞外信号调节激酶(extracellular signal-regulated kinase,ERK)、c-JunN 端激酶(c-Jun N terminal kinase,JNKs)和p38的复合物,它对基因诱导、细胞增殖和存活、细胞凋亡和分化、细胞应激和炎症反应等多种生物学结果产生重要影响,同时也是正常破骨细胞分化和激活的关键[30]。RANKL通过刺激下游信号MAPKs(ERK、JNK、p38)磷酸化激活,从而诱导了破骨细胞分化相关因子NFATc1的自我扩增;然后,NFATc1易位到细胞核,促进破骨细胞相关的TRAP、MMP9、CTSK、DC-STAMP和OSCAR基因的表达[15]。这一机制已在Xu等[31]的研究中得以证实,他们通过使用寡聚糖减弱RANKL介导的MAPK通路,发现NFATc1的表达降低,从而影响破骨细胞分化。ERK活化是成熟破骨细胞存活的关键[32]。Ras作为ERKs的上游激活蛋白,通过高亲和力和Raf-1 N-端结合,将Raf从胞浆转移到胞膜上,从而激活Raf在胞膜上的作用,进而诱导MAPK/ERK激酶MEK1和MEK2,从而控制ERK1和ERK2的激活;并导致多种下游效应物如c-Fos和NFATc1磷酸化[33]。NFATc1作为破骨细胞生成的重要转录因子,在ERK信号的调节下对破骨细胞的融合和骨吸收进行调控[34]。同样,JNKs也参与了破骨细胞的形成,由JNKs控制的AP-1和相关基因(c-Jun、JunB、c-Fos)对于破骨细胞的分化和成熟也是至关重要的[35]。RANKL诱导JNK活化,从而调节转录因子c-Jun的磷酸化,进而与c-Fos形成复合物,成为破骨细胞生成的关键转录因子;JNK信号也上调钙/钙调素依赖性蛋白激酶、c-Fos和NFATc1,因而促进形成破骨细胞[33]。Chang等[36]研究表明,炎症环境中的IL-16增强了JNK的磷酸化,进而激活下游分子NFATc1和TRAP,最终导致单核细胞分化为破骨细胞并导致小鼠骨质流失。这一研究表明,和ERK一样,JNK也通过影响NFATc1的转录影响骨平衡。此外,作为MAPK家族的一员,P38经RANKL介导从而激活下游c-Fos、NFATc 1等破骨细胞分化相关因子影响破骨分化[37]。已有研究者使用高姜素抑制了p38以及下游因子NFATc1、c-Jun和c-Fos的磷酸化可抑制破骨细胞的发生,从而有效地抑制骨质疏松症的发生和进展[38]。综上所述,作为影响破骨的重要下游信号通路之一,MAPK通路通过调节NFATc1影响破骨细胞的形成,而这一机制也将成为未来治疗骨质疏松症的重要靶点。
2.1.4Ca2+信号通路:除上述信号通路外,RANKL与RANK之间的相互作用还可以激活Ca2+通路影响破骨细胞[39],这对于破骨细胞增殖、分化、基因转录和骨吸收等多种功能至关重要[40]。前面章节已阐明在破骨细胞形成的早期阶段,RANK和RANKL的联合作用引发了TRAF6的激活并刺激PLCγ产生IP3,从而引起内质网Ca2+释放。细胞内骤升的Ca2+使通过CaM激活钙调磷酸酶后使NFAT调节结构域的丝氨酸残基去磷酸化,改变NFAT蛋白的构象,暴露出核定位序列,从而促进其从细胞质转移到细胞核,并在细胞核中作为转录因子进一步诱导和激活NFATc1[41],同时增加破骨细胞特异性基因(如TRAP、CTSK等)的表达,刺激破骨细胞前体的成熟,并增强破骨细胞相关蛋白的活性[42]。因此,Ca2+信号对于破骨细胞的分化和功能至关重要。Hong等[43]的研究结果发现,通过抑制RANKL诱导的Ca2+振荡的增加,从而抑制NFATc1的激活和自我扩增。这一研究结果表明抑制NFAT反式激活取决于RANKL诱导的Ca2+振荡的减少。另有研究发现,通过使用青蒿琥酯抑制TRAF6和下游PLCγ-1-Ca2+-NFATc1信号通路的表达可以减弱破骨细胞生成[44]。这些研究表明,破骨细胞转录因子NFATc1依赖于Ca2+信号通路,从而调节RANKL诱导的破骨细胞生成。
2.2 NFATc-1与骨质疏松
骨代谢过程中破骨细胞介导的骨吸收和成骨细胞介导的骨形成之间的平衡失调,是导致骨质疏松症发生的主要原因[45]。当破骨细胞对骨的吸收超过成骨细胞形成新骨的速度会损害骨骼稳态,导致骨质疏松症的持续发展[46]。
在破骨细胞的形成过程中,NFATc1作为“开关”启动破骨细胞的分化和成熟[9]。Long等[47]研究发现,通过抑制NFATc1进入细胞核来减少破骨细胞的产生和骨吸收,可用于治疗雌激素缺乏和破骨细胞亢进所致的骨质疏松症。另有研究表明,通过抑制NFATc1的表达水平和转录活性,可有效地抑制破骨细胞的形成和功能,从而进一步降低下游破骨细胞基因的表达水平[48]。这可能为研究针对破骨失衡诱导的骨质疏松症的治疗方法铺平道路。
NFATc1作为破骨细胞信号通路的重要下游转录因子之一,对其调节可以影响破骨细胞的生成,而这一机制也将有望成为未来治疗骨质疏松症的重要靶点。
2.3 NFATc-1与骨修复材料
随着组织工程技术的不断发展和完善,基于生物材料的治疗策略最近成为治疗骨质疏松症的有效替代方案[49]。已有研究人员探索钛植入物表面对破骨细胞发生的调控机制,通过评估不同钛植入物表面刺激后NFATc1的表达情况,他们发现仿生微/亚微层级表面可能通过下调NFATc1表达抑制破骨细胞分化,进而以促进种植体与骨的骨融合,从而提高骨质疏松患者的治疗效果[50]。Ha等[51]研究揭示了二氧化硅纳米颗粒在抑制RANKL刺激早期关键破骨细胞转录调节因子NFATc1方面的作用。在此基础上,他们进一步将此蛋白修饰于二氧化硅纳米颗粒表面并构建成具有抗骨质疏松活性的药物载体。这项研究揭示了纳米颗粒在骨代谢方面的潜在治疗应用。应用生物材料调控NFATc1从而抑制过度破骨吸收诱导的骨稳态失衡,这在未来将有望成为临床上治疗骨质疏松症的一种新的策略思想。
3 总结与展望
骨质疏松症是一种由破骨细胞介导的骨质吸收过程,其强度超过了成骨细胞介导的骨质形成,从而导致了骨平衡的破坏。既往研究证实,NFATc1可以通过调控RANKL-RANK、NF-κB、MAPK、Ca2+等信号通路的表达影响破骨细胞的生成与分化,在骨质疏松症领域发挥着重要作用。然而,对于NFATc1通过多种信号通路作用于破骨细胞生成过多诱导骨质疏松的相关机制研究较为零散。关于生物材料靶向NFATc1的研究还处于起步阶段,其机制和相关信号通路仍需进一步探索。
