利用反向遗传技术产生表达外源基因的重组SA11轮状病毒
2024-03-15柴萨萨刘夏飞宋敬东李金松裴银辉李利利段招军
柴萨萨,蔡 昊,刘夏飞,赵 静,宋敬东,李金松,裴银辉,李利利,段招军
A组轮状病毒(rotavirus A,RVA)是引起全世界婴幼儿急性重症胃肠炎和腹泻的主要病原体,每年相关的死亡病例达20多万例,其中多数发生在发展中国家,造成了巨大的社会经济负担[1-2]。RVA属于呼肠病毒目(Reovirales)平滑呼肠病毒科(Sedoreoviridea)轮状病毒属,是一种双链RNA病毒,其基因组包含11个节段,编码6种结构蛋白(VP1-4,VP6和VP7)和6种非结构蛋白(NSP1-6)[3]。
随着“反向遗传学”系统(reverse genetics system,RGS)的发展,研究者可在体外改造和拯救病毒,为基因功能研究和病毒拯救提供了强大的平台[4]。轮状病毒的反向遗传学系统最初于2006年建立,其依靠辅助病毒的帮助,基于部分质粒实现了RVA基因片段的替换[5]。之后经历10多年的不断探索,完全基于质粒的轮状病毒反向遗传系统在2017年建立[6]。近几年,利用反向遗传技术美国和日本报道多篇以轮状病毒作为载体表达外源蛋白的研究,这些研究主要以轮状病毒NSP1和NSP3片段为改造对象并插入外源基因。NSP1被认为是非必需病毒蛋白,具有拮抗干扰素的作用[7]。且有研究发现,牛轮状病毒A5-16株的NSP1片段(1 087 bp)ORF区缺失500 bp,只表达N端50个氨基酸,接种小鼠后仍能诱导腹泻[8]。这为基于轮状病毒NSP1截短片段作为载体表达外源基因提供思路。
本研究将A组轮状病毒SA11株的NSP1片段5′N端223至1 388的核苷酸截短(模拟牛A5-16株的缺失位点),并插入了终止-再启动元件(P2A)连接的EGFP基因,构建质粒pT7/SA11-NSP1-EGFP。采用已建立的“12质粒”轮状病毒反向遗传系统成功拯救具有良好遗传稳定性的重组病毒rSA11/NSP1-EGFP。旨在为进一步开发基于轮状病毒载体的联合疫苗研究提供理论依据。
1 材料与方法
1.1 主要试剂 DMEM培养基(Gibco),胎牛血清(FBS)(Gibco),胰蛋白示磷酸肉汤(TPB)(Sigma),非必需氨基酸溶液(NEAA)(Gibco),L-谷氨酰胺(L-Glutamine)(Sigma),青-链霉素(PS)(Gibco),胰蛋白酶(含EDTA)(Gibco),潮霉素(InvivoGen),质粒DNA大提试剂盒(QIAGEN),Opti-MEM(Gibco),转染试剂Trans IT○R-LT1(Mirusbio),猪九型胰蛋白酶(无EDTA)(Sigma),核酸提取试剂盒(Geneaid),SDS-PAGE凝胶制备试剂盒(Solarbio),快速银染试剂盒(碧云天生物技术),一步法RT-PCR试剂盒(Thermo Fisher)。
1.2 细胞培养 本研究使用的稳定表达T7 RNA聚合酶(T7 RNAP)的幼仓鼠肾细胞(BHK/T7-9)由中国疾病预防控制中心腹泻室刘夏飞博士赠送,培养条件为77%DMEM+10%FBS+10%TPB+1%NEAA+1%PS+1%L-Glutamine,每隔一代传代加潮霉素600 μg/mL以去除未表达T7 RNAP的细胞。非洲绿猴肾细胞(MA104)由本实验室保存,培养条件为89%DMEM+10%FBS+1%PS。培养环境均为37 ℃,5% CO2的细胞培养箱。
1.3 质粒构建 本研究使用的编码SA11-L2基因组的11个T7拯救质粒和pCMV/NP868R 辅助质粒由中国疾病预防控制中心腹泻室刘夏飞博士赠送。
根据文献报道构建截短NSP1片段中插入EGFP基因的质粒pT7/NSP1-EGFP-SA11:将SA11毒株NSP1片段5′端开始的第223~1 388位核苷酸删除,插入终止-再启动元件(P2A)连接的EGFP基因(720 bp),由苏州金唯智生物科技公司合成cDNA序列。合成的cDNA序列替换到原pT7/NSP1-SA11表达质粒的RsrII和NcoI位点来构建pT7/NSP1-EGFP-SA11质粒。构建质粒的核苷酸序列经直接测序证实。
转染所需的12个质粒按照质粒大提试剂盒QIAGEN EndoFree Plasmid Maxi Kit说明书提取,提取的质粒均经1.5%琼脂糖凝胶电泳鉴定条带大小符合超螺旋结构,并通过分光光度计测定其纯度和浓度,吸光度260/280≈1.9,每个质粒浓度均调整至1 mg/mL。
1.4 BHK/T7-9细胞转染及病毒拯救 采用“12质粒系统”进行病毒拯救[9],具体步骤如下:
BHK/T7-9细胞以4×105/孔的密度铺于六孔板,培养24 h至细胞汇合度达到80%左右进行质粒转染。转染时,首先将12个质粒混合,包括0.8 μg的pT7/NSP1-SA11、pT7/NSP3-SA11、pT7/NSP4-SA11、pT7/VP1-SA11、pT7/VP2-SA11、pT7/VP3-SA11、pT7/VP4-SA11、pT7/VP6-SA11、pT7/VP7-SA11、pCMV/NP868R,和2.4 μg的pT7/NSP2-SA11、pT7/NSP5-SA11,当拯救重组病毒rSA11/NSP1-EGFP时,将质粒pT7/NSP1-SA11替换成pT7/NSP1-EGFP-SA11。质粒混合物中加220 μL的Opti-MEM,用移液枪轻轻混匀,再加入40 μL的TransIT-LT1转染试剂,短暂涡旋混匀,置于室温下孵育20 min。孵育结束后,均匀滴入六孔板BHK/T7-9细胞中,轻晃摇匀,置于CO2培养箱继续培养。培养24 h后,换不含胎牛血清的培养基,加重悬的MA104细胞5×104/孔,4 h后每孔加胰蛋白酶至终浓度为0.5 μg/mL,混合培养4 d将细胞冻融3次后,14 000×g离心15 min,上清保存于-80 ℃,传代接毒备用。
1.5 拯救病毒传代培养 将MA104细胞以4×105/孔的密度接种于6孔板,培养2 d。细胞汇合成单层后,PBS清洗细胞2次,每孔加2 mL的DMEM,放培养箱继续培养。随后取离心后的病毒上清600 μL,加胰蛋白酶(15 μg/mL)、CaCl2(800 μg/mL)于37 ℃水浴锅活化病毒1 h,然后弃去6孔板中DMEM,均匀滴入活化后的上清液。放入培养箱吸附2 h,期间每20 min轻晃板子1次。吸附结束,弃去上清液,用PBS清洗2次,每孔加2 mL的DMEM(含胰蛋白酶5 μg/mL),继续培养,并每天观察细胞病变。按同样方法盲传3代来扩增病毒。
1.6 TCID50法测定病毒滴度 将MA104细胞以1×104/孔的密度铺于96孔板,培养1 d至细胞汇合成单层。取活化后的100 μL病毒上清做10倍稀释(10-1~10-9),以100 μL/孔分别接种至长满MA104细胞的96孔板中,对照采用活化处理的DMEM,每个稀释度和对照均做8个重复,放回培养箱吸附2 h。将病毒稀释液弃掉,用PBS清洗1次,每孔加150 μL含5 μg/mL胰蛋白酶的DMEM维持液,置于37 ℃、5% CO2的培养箱中培养7 d。每天观察并记录各稀释度出现CPE的孔数,然后使用Kaber法计算各病毒的TCID50值。
1.7 电镜观察 依照上述方法,将rSA11和rSA11/NSP1-EGFP接种于MA104细胞,MOI值为0.01,接种48 h后,吸取细胞培养液,2 000×g离心10 min,取上清,磷钨酸负染;细胞刮下收集,低速离心,去上清,加入2.5%等戊二醛固定,然后用1%四氧化锇固定,水洗3次,乙醇梯度脱水,用环氧树脂浸透,70 ℃聚合12 h。使用超薄切片机半手动获得超薄切片(80 nm),用醋酸铀酰和柠檬酸铅染色。最后,使用Tecnai12透射电子显微镜(FEI,荷兰)观察。
1.8 PAGE电泳分析 使用Virus Nucleic Acid Extraction Kit II(Geneaid,中国台湾)试剂盒提取病毒核酸。取核酸样品20 μL加10 μL 2×RNA上样缓冲液混匀后,在预制的1.5 mm的聚丙烯酰胺凝胶上(PAGE)电泳(5%浓缩胶和10%下层胶),80V跑30 min以浓缩样品后,70 V电泳6 h以分离条带。电泳结束后,采用银染试剂盒进行银染色,观察RVA的11条基因组并采用凝胶成像仪拍照。
1.9 RT-PCR鉴定 为进行重组病毒NSP1片段的鉴定,本研究设计了SA11病毒株的NSP1片段的特异性引物,SA11-NSP1-F: 5′-TGGATGCCAGTTCCTGATGC-3′和SA11-NSP1-R:5′-TGCCAGCTAGGCGCTACTCT-3′。首先按上述方法提取病毒核酸,经SuperScript II Reverse Transcriptase试剂盒(Invitrogen,美国)进行逆转录后,采用TAKARA的Premix Taq扩增rSA11/NSP1-EGFP(1 105 bp)与rSA11(1 459 bp)片段。反应条件:98 ℃ 15 s,55 ℃ 30 s,72 ℃ 1 min,35个循环。扩增产物送至北京天一辉远公司测序,比对分析rSA11和rSA11/NSP1-EGFP的NSP1基因片段序列。
1.10 生长动力学曲线 将拯救病毒按前述方法以MOI值为0.01接种于MA104细胞6孔板中,重组病毒(rSA11/NSP1-EGFP与rSA11)均做3个重复。在感染MA104细胞后0 h、12 h、24 h、36 h和48 h的时候分别收取培养液上清,14 000×g离心10 min ,上清-80 ℃保存。与此同时,取各时相培养液200 μL提取病毒核酸,参照毛彤瑶等人[10]建立的方法合成RotaA-NSP3的引物和探针,采用AgPath-IDTM One-step RT-PCR Kit试剂盒根据说明书进行Taqman Real-time PCR对上清中病毒进行定量,反应条件为45 ℃ 20 min,1循环;95 ℃ 10 min,1循环;95 ℃ 15 s,60 ℃ 1 min,40循环。计算各时相病毒的基因组拷贝数,绘制细胞内病毒RNA生长动力学曲线。
2 结 果
2.1 rSA11和rSA11/NSP1-EGFP的拯救 按照“12质粒”的轮状反向遗传系统,转染了12质粒的BHK/T7-9细胞与MA104细胞混合培养,在培养4 d后,取培养物上清首先采用轮状病毒胶体金初步检测,rSA11和rSA11/NSP1-EGFP上清均显示为轮状病毒阳性。将冻融离心后的培养物上清接种至MA104细胞进行病毒扩增,在显微镜下均观察到明显的细胞病变效应,具有典型的A组轮状病毒病变特征(图1A、B),即细胞变圆,胞质暗沉,细胞脱落。在对照组细胞中未观察到细胞病变效应(图1C)。
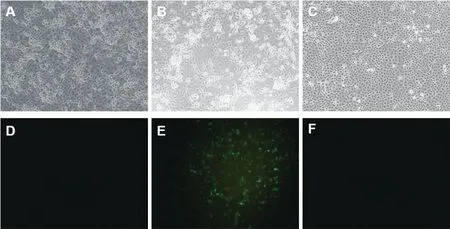
注:A、D. rSA11感染后细胞病变及荧光观察;B、E. rSA11/NSP1-EGFP感染后细胞病变及荧光观察;C、F.对照MA104细胞的细胞形态及荧光观察。
2.2 rSA11和rSA11/NSP1-EGFP的鉴定
2.2.1 rSA11和rSA11/NSP1-EGFP的荧光观察 将rSA11和rSA11/NSP1-EGFP的第3代培养物接种至MA104细胞。在荧光显微镜下观察,只在rSA11/NSP1-EGFP感染的MA104细胞中观察到明显的绿色荧光(图1E),在rSA11感染MA104细胞和对照孔细胞中并未观察到荧光(图1D、F)。
2.2.2 rSA11和rSA11/NSP1-EGFP的电镜观察 收集接种rSA11和rSA11/NSP1-EGFP的细胞培养上清,处理后,在透射电子显微镜下观察,均可观察到直径70 nm左右的球形轮状病毒粒子,包括实心和空心两种形态(图2)。
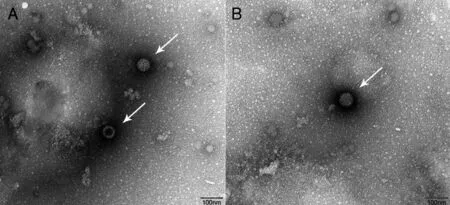
注:A.接种rSA11的细胞培养上清;B. 接种rSA11/NSP1-EGFP的细胞培养上清。
2.2.3 rSA11和rSA11/NSP1-EGFP的基因组鉴定 提取病毒核酸,进行了dsRNA基因组PAGE和RT-PCR鉴定。结果显示,rSA11/NSP1-EGFP与rSA11的11条基因节段有10条是一一对应的,而其中一条NSP1-EGFP迁移速度快于亲本株的NSP1片段(图3B)。RT-PCR的扩增产物经2%琼脂糖凝胶电泳鉴定,条带大小符合预期结果(图3A)。测序结果也显示,rSA11的NSP1片段与参考序列一致(1 459 bp),rSA11/NSP1-EGFP的NSP1片段包括完整的EGFP基因序列(1 178 bp)。这些结果表明我们成功拯救出了rSA11和插入外源基因的rSA11/NSP1-EGFP。
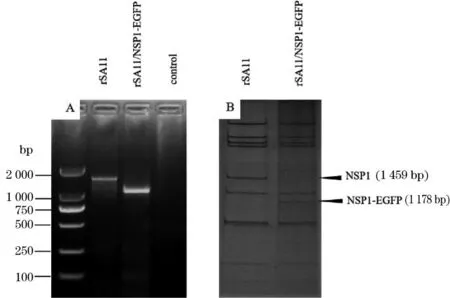
图3 rSA11和rSA11/NSP1-EGFP基因组的RT-PCR(A)和PAGE(B)鉴定Fig.3 RT-PCR (A) and PAGE (B) identification of rSA11 and rSA11/NSP1 EGFP genomes
2.3 重组病毒的滴度及生长动力学 为了确定NSP1截短的ORF区插入EGFP基因是否会影响重组病毒的感染力,取P3代病毒rSA11和rSA11/NSP1-EGFP以MOI为0.01感染MA104细胞,每隔12 h收取细胞培养液,定量不同时间点的RNA载量,绘制生长曲线。测定了48 h收取病毒液的TCID50。定量结果显示,rSA11和rSA11/NSP1-EGFP在MA104细胞中复制良好,并产生明显的CPE。rSA11和rSA11/NSP1-EGFP均在12 h已开始复制,在36 h到达复制平台期,48 h与36 h的病毒拷贝数无差异,两个毒株的复制动力学趋势一致(图4B)。Real-time PCR 和TCID50结果显示,rSA11/NSP1-EGFP的病毒RNA水平和病毒滴度略低于rSA11,P<0.05,差异具有统计学意义(图4A和4B)。
2.4 重组病毒的遗传稳定性 为验证重组病毒是否可以稳定传代,取P5和P10代重组病毒rSA11/NSP1-EGFP进行dsRNA基因组PAGE分析, rSA11作为对照。结果显示P5和P10代病毒rSA11/NSP1-EGFP的NSP1-EGFP片段条带位置一致,均出现正确迁移(图5)。结果表明,将NSP1片段ORF区截短并插入720 bp的EGFP基因,获得重组病毒的遗传稳定性在连续传代10次后依旧保持基因稳定性。
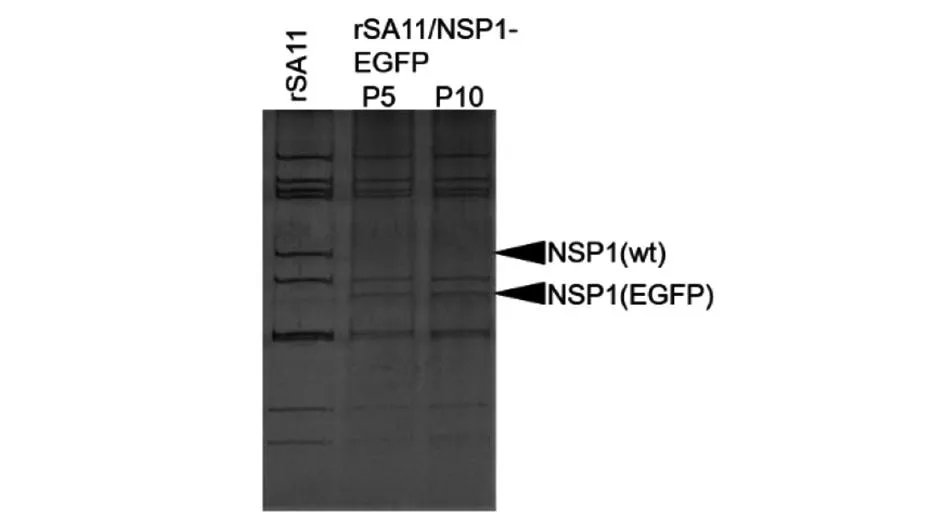
图5 rSA11/NSP1-EGFP的dsRNA遗传稳定性Fig.5 Genetic stability of dsRNA in rSA11/NSP1 EGFP
3 讨 论
A组轮状病毒仍是全世界婴幼儿严重急性胃肠炎的最重要原因。针对轮状病毒感染目前尚无特异性药物,主要采取对症治疗,疫苗接种是预防RV感染的有效措施。虽然十多年前开始在全球引入了轮状病毒疫苗接种,如:Rotarix、RotaTeq、Rotavac、ROTASIIL、Rotavin-M1、LLR,但轮状病毒感染每年仍导致全球20万人以上死亡,主要是在低收入国家[11]。人用商品化RV疫苗在发达国家有较高免疫效率,而在低收入国家免疫效率较低,且存在一定的潜在安全问题[12]。因此,对于新生儿、老年人和免疫缺陷的成年人来说开发安全并高效的抗 RV 的药物和疫苗都是十分迫切的。
反向遗传学是研究宿主-病原体相互作用、病毒蛋白质功能和病毒感染生物学的有力工具,且在新型疫苗设计方面有很大潜力[13-15]。轮状病毒反向遗传学系统的建立较为滞后,于2017年建立完全基于质粒的14质粒RVA反向遗传系统[6]。之后不久,Philip AA等人通过修改其程序为12质粒系统,进一步提高了病毒拯救效率。该系统通过将11个T7驱动的编码RVA基因组的质粒和1个编码非洲猪瘟病毒加帽酶的辅助质粒(pCMV/NP868R)借脂质体转染到表达T7 RNA聚合酶的BHK/T7-9细胞,重组轮状病毒可以高效获得[9]。最新研究表明,仅转染11个编码RVA基因组的拯救质粒,仍可高效拯救重组病毒[16]。基于这3种轮状病毒反向遗传系统,研究人员成功的产生了多种重配以及表达报告基因的具有感染性的重组轮状病毒[16-18]。
我们在拯救rSA11病毒株时对拯救过程进行了一些改进,经验如下:①最关键的是状态良好的BHK/T7-9细胞,使用添加10%胎牛血清、1%NEAA、10%TPB的高糖DMEM完全培养基,额外的营养成分补充有助于BHK/T7-9细胞在质粒转染后延长活力,此外细胞每周2次传代,隔代使用潮霉素补充培养基,用于筛选稳定表达T7 RNA聚合酶的BHK/T7-9细胞;②最佳的细胞密度,即只在细胞汇合度达到80%时进行转染;③转染试剂和培养基在使用前要平衡至室温;④转染时可适当增加11个T7拯救质粒的用量1~2倍;⑤转染前要确保质粒和转染试剂充分混合;⑥加入MA104细胞混合培养期间按需添加碳酸氢钠控制细胞培养的pH值。另外,支原体污染BHK/T7-9和MA104细胞可能是轮状病毒反向遗传系统出现重组病毒失败的重要因素。在细胞的培养过程中,我们同时使用基于PCR和荧光染色法的支原体检测试剂盒定期来检查BHK/T7-9细胞和MA104细胞,确保用到的细胞无支原体污染,如若检测到,重新复苏新的细胞系,丢弃之前使用过的培养基和培养基补充剂,并彻底清洗细胞培养箱,生物安全柜,实验室工作台和移液器。
本研究将轮状病毒SA11株的NSP1片段5′端223至1 388的核苷酸截短,并插入了P2A终止-再启动元件连接的EGFP基因,构建质粒pT7/SA11-NSP1-EGFP。采用已建立的RV反向遗传12质粒系统[9],以pT7/SA11-NSP1-EGFP质粒替换掉原来系统的pT7/SA11-NSP1质粒,成功拯救出了表达荧光报告基因(EGFP)的具有感染性的重组轮状病毒,并通过RT-PCR、SDS-PAGE电泳、电镜观察及间接免疫荧光等方法,进行了一系列的结果验证,证明了RVAs除了其原始基因组dsRNA外,还可以携带外源核苷酸作为遗传物质,而且连续10次传代后重组病毒基因组依然保持稳定。当我们按相同MOI值接种病毒时,重组病毒的复制效率略低于亲本株,且拯救效率也要低于亲本株。下一步的工作重点将提高重组病毒的拯救效率。
在重组RVA具有感染性的情况下,荧光报告基因的表达使得RVA在体内和体外复制实现可视化。Hatazawa R等人已报告基于该系统可以生成表达多种外源基因的重组RV[19],插入片段的长度可达2 160 bp,这使其成为一种有潜力的灵活病毒载体。事实上,可以将编码其他病原体中和抗原的序列插入RVA基因组,从而产生基于RVA的多价载体疫苗[20-21],不仅可以诱导对RVA的免疫保护,还可以诱导对插入其他病原体的免疫保护。因此,基于RVA的载体将在广泛的基础研究和临床应用中具有吸引力。
利益冲突:无
引用本文格式:柴萨萨,蔡昊,刘夏飞,等.利用反向遗传技术产生表达外源基因的重组SA11轮状病毒[J].中国人兽共患病学报,2024,40(2):140-146. DOI:10.3969/j.issn.1002-2694.2024.00.020
