液相色谱--串联质谱法测定茶叶中敌百虫的不确定度评定
2024-03-08杨丽蓉段联勃柯顺川符海霞
杨丽蓉 段联勃,2 柯顺川 符海霞
(1.武夷星茶业有限公司,福建武夷山,354300;2.福建省企业技术中心,福建武夷山,354300)
日常的测量过程中,受到人员重复性、器皿设备精密度、试剂浓度精确度、方法适宜性及环境条件波动等因素的影响,测量值往往在一个区间内波动。不确定度反映了测量值的分散程度。不确定度的评定能够帮助人们了解这种波动的程度,掌握测量值的可信程度。通过对影响测量值的不同因素进行不确定度评定,发现主要的不确定度分量,从而寻找出测量方法的改进机会,缩小波动的范围进而增加测量值的可信度。
敌百虫是一种高效、速效、广谱,低残留的有机磷农药,有熏蒸、胃毒和触杀等功能,可高效防治稻米、小麦、蔬菜、茶树、水果、桑树以及棉花等经济作物上的咀嚼式口器害虫、家畜寄生虫和卫生害虫等[1]。GB2763-2021中规定茶叶中敌百虫最大残留量为2mg/kg,但茶叶样品基质复杂,含有生物碱、色素、茶多酚等一系列干扰成分,给敌百虫的准确测定造成了较大的不确定性[2]。在现有的茶叶检测中,不确定度评定的文献中以农药残留[5-8]和重金属[9-12]为主,但有关液相色谱-串联质谱法(Liquid chromatography - mass spectrometry,LC-MS/MS)检测茶叶中敌百虫的不确定度评定研究较为少见。
本研究以JJF 1059.1-2012《测量不确定度评定与表示》和CNAS-GL006-2019《化学分析中不确定度的评估指南》为依据,分析和评定了GB 23200.13-2016测定茶叶中敌百虫残留量的不确定度,为评价茶叶中敌百虫残留量测定结果的准确性与可靠性提供了一定的技术依据。
1 材料与方法实验部分
1.1 试验材料
乙腈、甲苯:色谱纯,西陇科学股份有限公司;甲醇:色谱纯,天津市光复科技发展有限公司;CleanertTPT 固相萃取柱:10mL,2g,博纳艾杰尔公司;微孔滤膜(尼龙):13mm×0.2um,江苏绿盟科学仪器有限公司;敌百虫标准物质:纯度97. 2%,德国Dr.Ehrenstorfer 公司;实验用水为优普纯水机制备的超纯水:电阻率
1.2 试验方法
1.2.1 标准溶液配制
标准储备液(500mg/L):准确称取折算纯度后的敌百虫标准物质25.72mg,加甲醇溶解定容至50mL。
标准中间液(50mg/L):用5mL 移液器移取2.5mL 标准储备液至25mL 容量瓶中,加甲醇定容至刻度。
工作标准溶液(1.0mg/L):用1mL 移液器移取中间液0.5mL 至25mL 容量瓶中,加甲醇定容至刻度。
上机标液配制:用1mL 移液器分别移取100uL、200uL、400uL、800uL、1000uL 工作溶液,至5mL 容量瓶中,空白基质提取液定容至刻度,移取1mL 上机。
1.2.2 样品测定
称取1 0.0 g 试样(精确至0.0 1 g)于5 0 m L 具塞离心管中,加入3 0 m L 乙腈溶液,以1 5 0 0 0 r/m i n 均质提取1 m i n,4 2 0 0 r/m i n 离心5 m i n,移取上清液至鸡心瓶中。残渣加3 0 m L乙腈,重复上述均质离心过程,将上清液合并至鸡心瓶中;再向残渣中加入2 0 m L 乙腈,重复均质离心过程,合并上清液至同一鸡心瓶中,4 5 ℃旋转蒸发至近干,氮吹至干。残留物用5 m L 乙腈复溶待净化。C l e a n e r t T P T 加入约2 c m 高无水硫酸钠后,加入5 m L 乙腈-甲苯溶液,待液面到达无水硫酸钠顶部时,迅速移取1 m L 复溶液至C l e a n e r t T P T 净化柱上,并更换另一洁净鸡心瓶接收。用2 5 m L 乙腈-甲苯溶液洗脱,收集洗脱液于鸡心瓶中,4 5 ℃水浴浓缩至约0.5 m L,转至3 5 ℃下氮气吹干,加入1 m L 乙腈-水溶液溶解残渣,经微孔滤膜过滤后,供液相色谱-串联质谱测定。进行平行试验,采用标准曲线法定量,测定结果用平行测定的算术平均值表示。
1.3 测量不确定度的评定方法
1.3.1 数学模型
式中:
X—茶样中敌百虫的含量,单位:mg/kg;
C—标准工作曲线中得到的敌百虫浓度,单位:mg/L;
V—样液定容体积,单位:mL;
M—试样质量,单位:g;
REP—总重复因子;
R—敌百虫回收率;
f—样品稀释因子,f=5。
1.3.2 不确定度来源的确定和分析
不确定度来源:测量重复性引入的不确定度分量ur(REP);检测样品称量过程引入的不确定度分量ur(M);仪器测定带来的不确定度分量ur(LCMSMS);标准溶液配制及稀释时引入的不确定度分量ur(C);标准曲线拟合引起的不确定度分量ur(L);样液定容体积引起的不确定度分量ur(V);回收率带来的不确定度分量ur(R);样品的制备和分样严格按照检测标准要求执行,因此样品均匀性引入的不确定度可忽略不计[18]。由数学模型导出:
2 结果与分析
2.1 不确定度的评定
2.1.1 测量重复性带来的不确定度分量ur(REP)
该分量含人员、环境、测量方法及仪器重复性等其它分量的贡献。样品加标进行10 次重复分析,加标浓度为0.03mg/kg。测量结果通过下式计算重复性引入的相对标准不确定度ur(REP),测量及计算结果见表1。
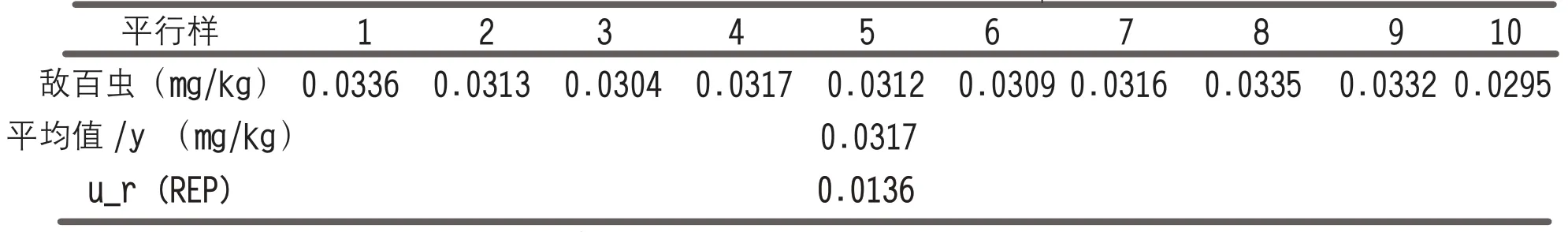
表1 测量重复性引入的相对标准不确定度u (REP)
式中:
u(REP)—重复分析的标准不确定度;
yi—第i 个平行样的测量结果;
y —测量结果的平均值;
n—平行样的个数,n=10;
ur(REP)—重复性引入的相对标准不确定度。
样品称量时使用的电子天平精度为0.01g,每次样品称量均准确称取10.00g,电子天平校准证书显示称量范围的最大允许误差为±0.05g,依据矩形分布(k=√3),则:
样品称量的标准不确定度u(M)=δ/√3=0.05/√3=0.0289(g),
试样质量为10.00g,其示值引入的相对标准不确定度ur(M)=0.0289/10.00=0.0029。
2.1.3 液相色谱质谱联用仪仪器测定带来的不确定度分量ur(LCMSMS)
计量证书显示定量重复性为1%,依据矩形分布,该设备定量重复性引入的相对标准不确定度:
2.1.4 标准溶液配制和稀释过程引入的不确定度分量ur(C)
该不确定度分量包括标准物质纯度引起的标准不确定度ur(p)、标准物质称量引起的标准不确定度ur(m)、操作温度引入的不确定度ur(T),实际操作与校正温度相同,则温度引入的不确定度可忽略、储备液配制定容到50mL 引起的标准不确定度ur(V1)、配制中间标准液移取储备液引起的标准不确定度ur(V2)、中间标准液定容引起的标准不确定度ur(V3)、配制工作溶液移取中间液引起的标准不确定度ur(V4)、工作溶液定容引起的标准不确定度ur(V5)、移取不同体积工作溶液配制工作曲线引起的标准不确定度ur(V6)、标准工作曲线定容引起的标准不确定度ur(V7),则有:
标准物质证书显示,敌百虫标准物质的扩展不确定度U=1.02%,纯度p 为97.20%,包含因子k=2,则下式计算出标准物质纯度带入的相对标准不确定度ur(p)=0.00525。
称量使用精度为0.01mg 的分析天平,每次称量均准确称取经纯度折算后的质量,电子天平校准证书显示,称量范围的最大允许误差为±0.5mg,依据矩形分布(K=√3),则标准物质称量带入的相对标准不确定度ur(m)=0.5/(√3×25/p)=0.01123。
50mL 容量瓶校准证书显示,容量瓶最大允误差为±0.05mL,依据矩形分布(k=√3),则储备液配制定容到50mL 带入的相对标准不确定度ur(V1)=0.05/(√3×50)=0.00058。
配制中间标准溶液用5mL 移液器移取储备液2.5mL,查5mL 移液器校准证书移取范围的最大允许误差为±0.5%,依据矩形分布(K=√3),则配制中间标准液移取储备液引起的相对标准不确定度ur(V2)=0.5%×2.5/(√3×2.5)=0.00289。
中间标准溶液定容至25mL,根据25mL 容量瓶校准证书提供的信息,容量瓶最大允许误差为±0.03mL,依据矩形分布(k=√3),则该定容过程引起的相对标准不确定度ur(V3)=0.03/(√3×25)=0.00070。
配制工作标准液移取中间液用1mL 移液器移取中间液0.5mL,查1mL 移液器校准证书移取范围的最大允许误差为±1.0%,依据矩形分布(k=√3),则该移取过程引起的相对标准不确定度ur(V4)=1.0%×0.5/(√3×0.5)=0.00578。
工作标准溶液定容至25mL,25mL 容量瓶校准证书显示,25mL 容量瓶最大允差为±0.03mL,依据矩形分布(k=√3),则该定容过程引起的相对标准不确定度ur(V5)=0.03/(√3×25)=0.00070。
配制上机工作曲线标液移取过程使用1mL 的移液器移取100μL、200μL、400μL 、800μL、1000μL共5 次,查1mL 移液器校准证书在移取范围小于500μLuL 的最大允许误差为±2.0%,按矩形分布(k=√3),则单次不确定度为:
在移取范围大于500μL的最大允许误差为±1.0%,则单次不确定度为:ur(V800μL)=ur(V1000μL)=1.0%/√3=0.00578;
配制上机工作曲线标液移取过程引起的标准不确定度ur(V6)通过下式结算结果为0.02162。
配制上机工作曲线标液定容过程,各曲线点定容至5mL,共定容5 次,查5mL 容量瓶校准证书,5mL 容量瓶最大允许误差为±0.02mL,依据矩形分布(k=√3),则单次不确定度为:u(V7)=0.02/(√3×5)=0.00231
则配制上机工作曲线标液定容过程引起的相对标准不确定度为:ur(V7)=√(5×u(V7)2=0.00517
则:标准溶液配制和稀释时带入的相对不确定度ur(C)通过下式计算,ur(C)=0.0263。
2.1.5 标准曲线拟合引入的不确定度分量ur(L)
采用混合标液稀释,用空白基质配制成5 种相对含量的标准溶液(10μg/kg、20μg/kg、40μg/kg、80 μg/kg、100μg/kg),每种浓度测定1 次,上机分别测定,每个浓度点测1 次,获得相应峰面积,以最小二乘法拟合,得到直线方程y=bx+a(a 为截距,b 为斜率)和相关系数r,并按下面公式计算出拟合线性的标准差SL和拟合曲线引入的相对不确定度ur(L),具体数据及计算结果见表2。拟合线性的标准差按下式计算:
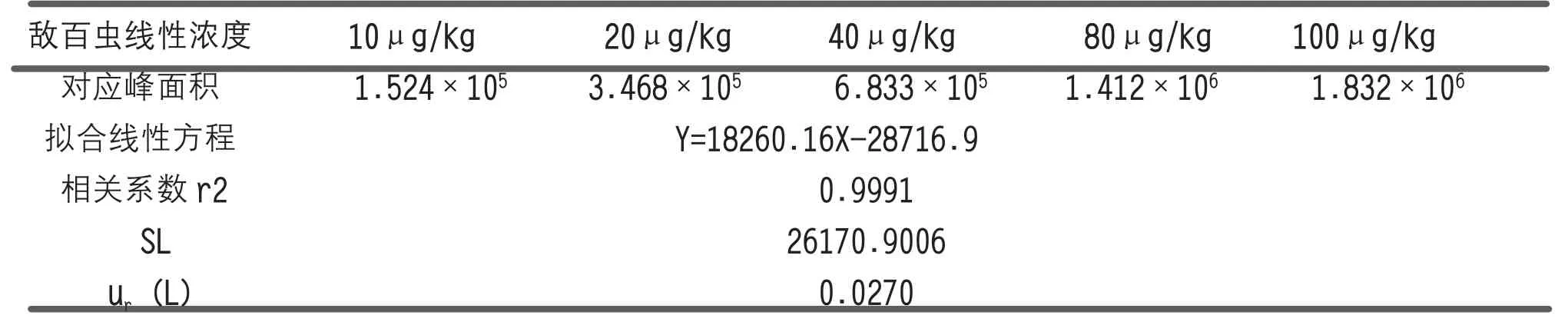
表2 标准曲线拟合引入的相对标准不确定度分量ur(L)
式中:
yij—农药及相关化学品各浓度点的面积;;
m—每个浓度点的测量次数;1 次
n—线性浓度点个数;
SL—拟合曲线的标准差。
拟合曲线引入的不确定度u(L)按下式计算:
式中:
S_L—拟合曲线的标准差;
b—拟合曲线的斜率;
P—测试样品的次数;
n—测定标准溶液的次数(各浓度点之和);
x—未知样的含量,单位μg/kg;
xi—标准曲线每个点对应含量,单位μg/kg。
则拟合曲线引入的相对不确定度ur(L)按下式计算:
2.1.6 样液定容体积引起的不确定度分量ur(V)
用1mL(100-1000μL)移液器移取定容,其最大允许误差为1.0%,依据矩形分布,k=√3,则:
吸取1mL 乙腈5 次溶解残渣,则单次相对标准不确定度为:ur(V乙腈)=1.0%/√3=0.00578;
移取1mL 溶解物萃取时的相对标准不确定度为:ur(V溶解物)=1.0%/√3=0.00578;
吸取1mL 乙腈水时的相对标准不确定度为:ur(V乙腈水)=1.0%/√3=0.00578
则样液定容过程引入的相对标准不确定度为:
2.1.7 回收率校正结果引起的不确定度分量ur(R)
表3 收集了汇总了近10 次日常添加的回收率及均值数据,1、2 组添加浓度为0.03mg/kg;3、4、5、6 组添加浓度为0.06mg/kg;7、8、9、10 组添加浓度为0.15mg/kg。回收率带入的相对标准不确定度ur(R)通过下面两式计算,计算结果见表3:
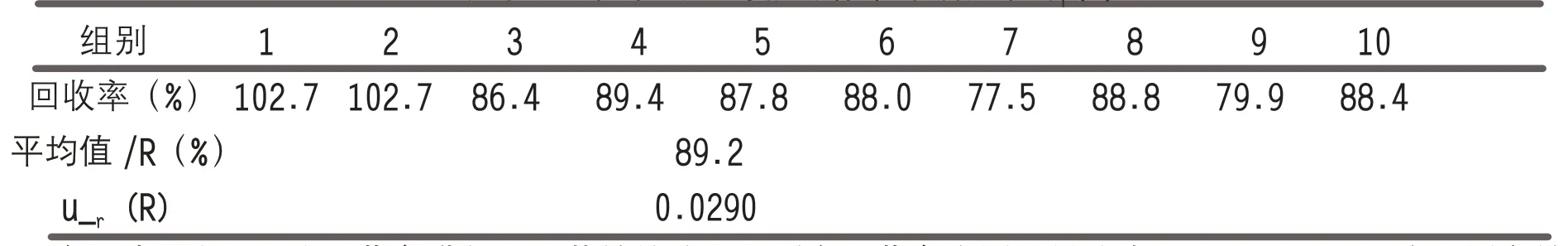
表3 回收率引入的相对标准不确定度u (R)
式中:
u(R)—回收率引起的标准不确定度;
Ri—第i 组测定的回收率结果;
R —回收率的平均值;
n—组别的个数,n=10;
ur(R)—回收率带入的相对标准不确定度。
并通过下式对平均回收率进行t 显著性检验,以判定回收率在测量模式中是否需要采用以修正测定结果。在95%置信区间,自由度为9 时,t 分布临界值为2.262,当t 检验值大于等于该临界值时,说明存在显著性差异,需要使用回收率进行结果校正,否则不需要。经计算检验值t=3.724,需要进行回收率校正。
2.2 合成标准不确定度的计算
按照以下公式可得合成标准不确定度为,经计算合成标准不确定度ur(X)=0.0522。
2.3 扩展不确定度U 的计算
取95%置信水平,k=2,敌百虫含量测定的扩展不确定度根据以下公式计算,结果为U=0.0034。
2.4 不确定度报告
按照根据GB 23200.13-2016 规定的检测程序进行,本次茶叶中敌百虫含量测定的结果表示如下:敌百虫含量=(0.0317±0.0034)mg/kg; k=2。
2.5 结语贡献率
根据以下公式计算敌百虫含量测定不确定度各分量的贡献率根据以下公式计算,结果见表4。
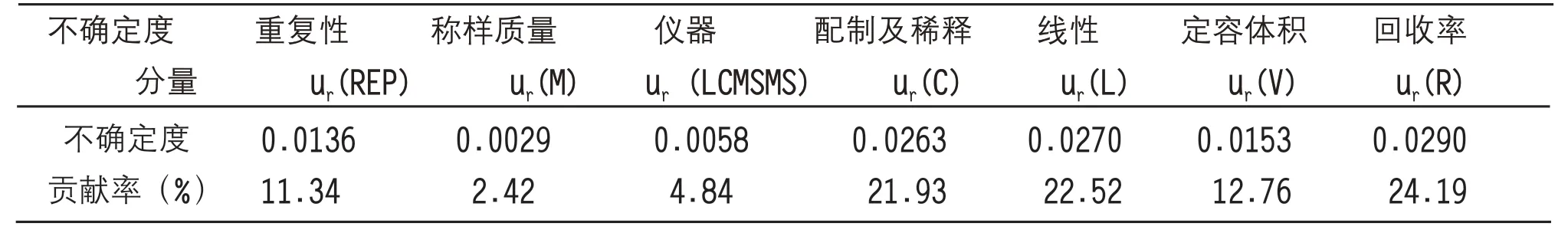
表4 不确定度分量的贡献率
如表4 所示,通过比较不确定度各个分量的贡献率显示,敌百虫检测过程各环节对检测结果的不确定度影响程度不同。过程中影响最大的是回收率、标准曲线最小二乘法拟合和标准溶液配制与稀释过程的分量,其余因素按照从大到小依次为定容体积、测量重复性、液相色谱质谱联用仪LC-MS/MS 仪器测定、样品称量质量。因此,按该标准进行敌百虫检测分析时,可通过将检测结果进行回收率校正、加大标准曲线各浓度点的测定次数、使用精确度更高的移液管或其它移取设备进行标样配制和定容以及增加检测样品平行测定的次数等手段来减少这些分量引入的不确定度,提高检测结果的可靠性。
3 结论与讨论
本文使用液相色谱-串联质谱法对茶叶中敌百虫的残留量进行了分析,通过数学模型对不确定度的来源进行分析和确定后,分别对各不确定度分量进行了评定,最后通过不确定度分量贡献率显示:该方法中回收率带入的不确定分量为最大不确定度。贡献率的分析能够为方法的优化提供方向,从而通过合理优化提高检测的准确性。文章描述了不确定度评定的过程,为评价茶叶中农药残留测定的不确定度评定提供了一定的参考价值。

